 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImageDDI: Image-enhanced Molecular Motif Sequence Representation for Drug-Drug Interaction Prediction
Aug 11, 2025To mitigate the potential adverse health effects of simultaneous multi-drug use, including unexpected side effects and interactions, accurately identifying and predicting drug-drug interactions (DDIs) is considered a crucial task in the field of deep learning. Although existing methods have demonstrated promising performance, they suffer from the bottleneck of limited functional motif-based representation learning, as DDIs are fundamentally caused by motif interactions rather than the overall drug structures. In this paper, we propose an Image-enhanced molecular motif sequence representation framework for \textbf{DDI} prediction, called ImageDDI, which represents a pair of drugs from both global and local structures. Specifically, ImageDDI tokenizes molecules into functional motifs. To effectively represent a drug pair, their motifs are combined into a single sequence and embedded using a transformer-based encoder, starting from the local structure representation. By leveraging the associations between drug pairs, ImageDDI further enhances the spatial representation of molecules using global molecular image information (e.g. texture, shadow, color, and planar spatial relationships). To integrate molecular visual information into functional motif sequence, ImageDDI employs Adaptive Feature Fusion, enhancing the generalization of ImageDDI by dynamically adapting the fusion process of feature representations. Experimental results on widely used datasets demonstrate that ImageDDI outperforms state-of-the-art methods. Moreover, extensive experiments show that ImageDDI achieved competitive performance in both 2D and 3D image-enhanced scenarios compared to other models.
Y-Mol: A Multiscale Biomedical Knowledge-Guided Large Language Model for Drug Development
Oct 15, 2024Large Language Models (LLMs) have recently demonstrated remarkable performance in general tasks across various fields. However, their effectiveness within specific domains such as drug development remains challenges. To solve these challenges, we introduce \textbf{Y-Mol}, forming a well-established LLM paradigm for the flow of drug development. Y-Mol is a multiscale biomedical knowledge-guided LLM designed to accomplish tasks across lead compound discovery, pre-clinic, and clinic prediction. By integrating millions of multiscale biomedical knowledge and using LLaMA2 as the base LLM, Y-Mol augments the reasoning capability in the biomedical domain by learning from a corpus of publications, knowledge graphs, and expert-designed synthetic data. The capability is further enriched with three types of drug-oriented instructions: description-based prompts from processed publications, semantic-based prompts for extracting associations from knowledge graphs, and template-based prompts for understanding expert knowledge from biomedical tools. Besides, Y-Mol offers a set of LLM paradigms that can autonomously execute the downstream tasks across the entire process of drug development, including virtual screening, drug design, pharmacological properties prediction, and drug-related interaction prediction. Our extensive evaluations of various biomedical sources demonstrate that Y-Mol significantly outperforms general-purpose LLMs in discovering lead compounds, predicting molecular properties, and identifying drug interaction events.
Conditioned Generative Transformers for Histopathology Image Synthetic Augmentation
Dec 20, 2022



Deep learning networks have demonstrated state-of-the-art performance on medical image analysis tasks. However, the majority of the works rely heavily on abundantly labeled data, which necessitates extensive involvement of domain experts. Vision transformer (ViT) based generative adversarial networks (GANs) recently demonstrated superior potential in general image synthesis, yet are less explored for histopathology images. In this paper, we address these challenges by proposing a pure ViT-based conditional GAN model for histopathology image synthetic augmentation. To alleviate training instability and improve generation robustness, we first introduce a conditioned class projection method to facilitate class separation. We then implement a multi-loss weighing function to dynamically balance the losses between classification tasks. We further propose a selective augmentation mechanism to actively choose the appropriate generated images and bring additional performance improvements. Extensive experiments on the histopathology datasets show that leveraging our synthetic augmentation framework results in significant and consistent improvements in classification performance.
Nearest Descent, In-Tree, and Clustering
Jan 25, 2018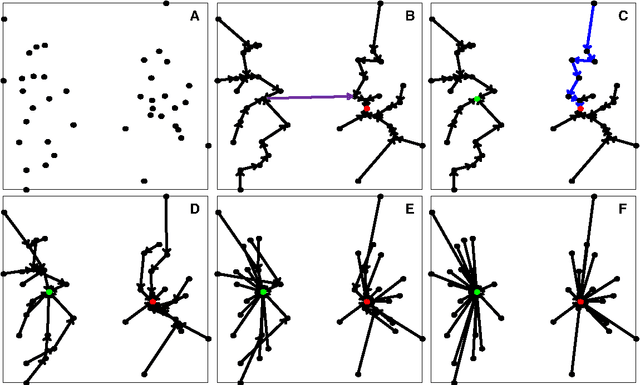
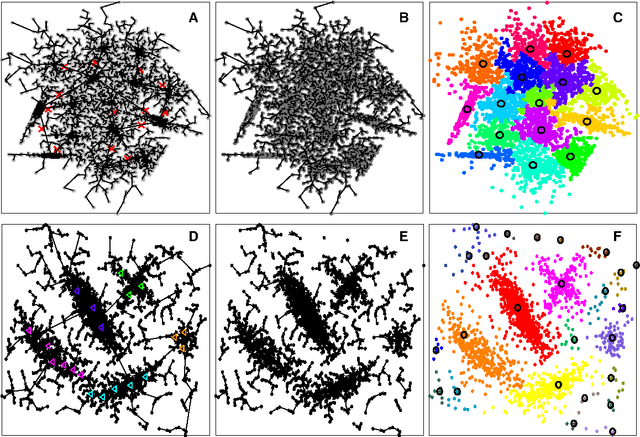
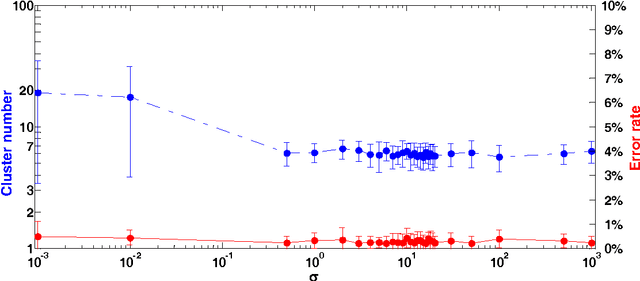

In this paper, we propose a physically inspired graph-theoretical clustering method, which first makes the data points organized into an attractive graph, called In-Tree, via a physically inspired rule, called Nearest Descent (ND). In particular, the rule of ND works to select the nearest node in the descending direction of potential as the parent node of each node, which is in essence different from the classical Gradient Descent or Steepest Descent. The constructed In-Tree proves a very good candidate for clustering due to its particular features and properties. In the In-Tree, the original clustering problem is reduced to a problem of removing a very few of undesired edges from this graph. Pleasingly, the undesired edges in In-Tree are so distinguishable that they can be easily determined in either automatic or interactive way, which is in stark contrast to the cases in the widely used Minimal Spanning Tree and k-nearest-neighbor graph. The cluster number in the proposed method can be easily determined based on some intermediate plots, and the cluster assignment for each node is easily made by quickly searching its root node in each sub-graph (also an In-Tree). The proposed method is extensively evaluated on both synthetic and real-world datasets. Overall, the proposed clustering method is a density-based one, but shows significant differences and advantages in comparison to the traditional ones. The proposed method is simple yet efficient and reliable, and is applicable to various datasets with diverse shapes, attributes and any high dimensionality