 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMedAttacker: Exploring Black-Box Adversarial Attacks on Risk Prediction Models in Healthcare
Dec 11, 2021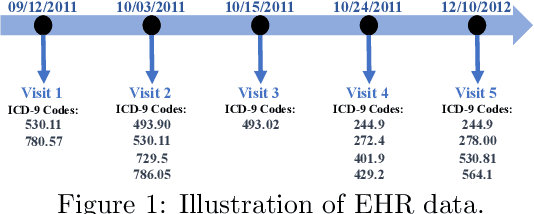
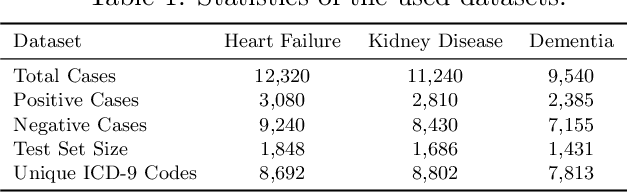
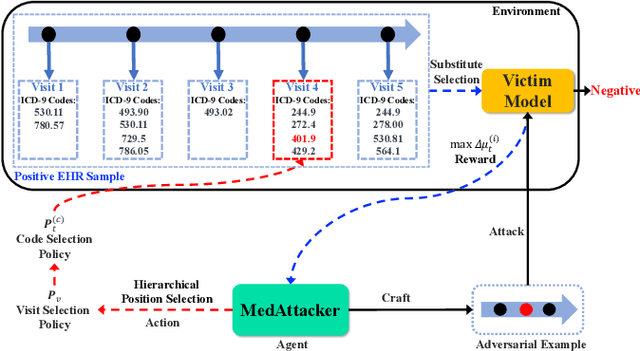
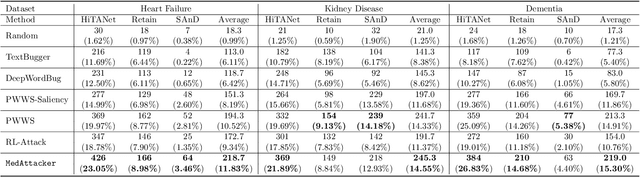
Deep neural networks (DNNs) have been broadly adopted in health risk prediction to provide healthcare diagnoses and treatments. To evaluate their robustness, existing research conducts adversarial attacks in the white/gray-box setting where model parameters are accessible. However, a more realistic black-box adversarial attack is ignored even though most real-world models are trained with private data and released as black-box services on the cloud. To fill this gap, we propose the first black-box adversarial attack method against health risk prediction models named MedAttacker to investigate their vulnerability. MedAttacker addresses the challenges brought by EHR data via two steps: hierarchical position selection which selects the attacked positions in a reinforcement learning (RL) framework and substitute selection which identifies substitute with a score-based principle. Particularly, by considering the temporal context inside EHRs, it initializes its RL position selection policy by using the contribution score of each visit and the saliency score of each code, which can be well integrated with the deterministic substitute selection process decided by the score changes. In experiments, MedAttacker consistently achieves the highest average success rate and even outperforms a recent white-box EHR adversarial attack technique in certain cases when attacking three advanced health risk prediction models in the black-box setting across multiple real-world datasets. In addition, based on the experiment results we include a discussion on defending EHR adversarial attacks.
Differentiable Scaffolding Tree for Molecular Optimization
Sep 22, 2021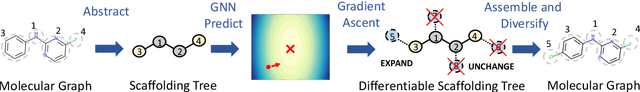

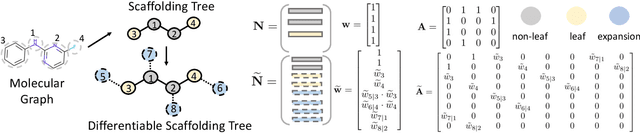
The structural design of functional molecules, also called molecular optimization, is an essential chemical science and engineering task with important applications, such as drug discovery. Deep generative models and combinatorial optimization methods achieve initial success but still struggle with directly modeling discrete chemical structures and often heavily rely on brute-force enumeration. The challenge comes from the discrete and non-differentiable nature of molecule structures. To address this, we propose differentiable scaffolding tree (DST) that utilizes a learned knowledge network to convert discrete chemical structures to locally differentiable ones. DST enables a gradient-based optimization on a chemical graph structure by back-propagating the derivatives from the target properties through a graph neural network (GNN). Our empirical studies show the gradient-based molecular optimizations are both effective and sample efficient. Furthermore, the learned graph parameters can also provide an explanation that helps domain experts understand the model output.
Augmented Tensor Decomposition with Stochastic Optimization
Jul 14, 2021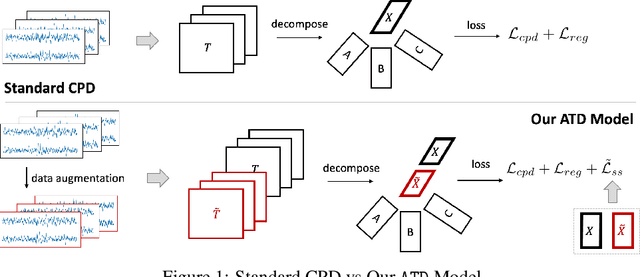
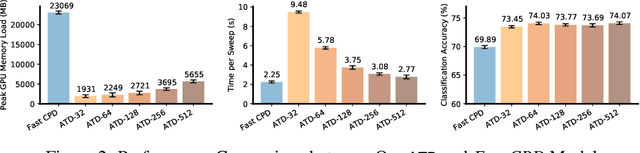
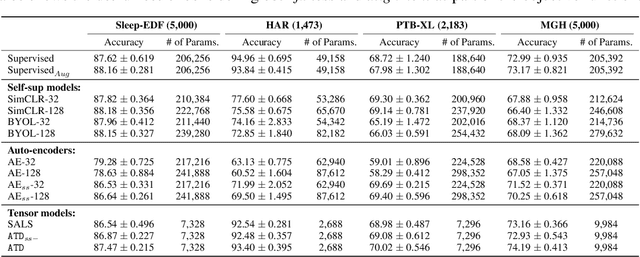
Tensor decompositions are powerful tools for dimensionality reduction and feature interpretation of multidimensional data such as signals. Existing tensor decomposition objectives (e.g., Frobenius norm) are designed for fitting raw data under statistical assumptions, which may not align with downstream classification tasks. Also, real-world tensor data are usually high-ordered and have large dimensions with millions or billions of entries. Thus, it is expensive to decompose the whole tensor with traditional algorithms. In practice, raw tensor data also contains redundant information while data augmentation techniques may be used to smooth out noise in samples. This paper addresses the above challenges by proposing augmented tensor decomposition (ATD), which effectively incorporates data augmentations to boost downstream classification. To reduce the memory footprint of the decomposition, we propose a stochastic algorithm that updates the factor matrices in a batch fashion. We evaluate ATD on multiple signal datasets. It shows comparable or better performance (e.g., up to 15% in accuracy) over self-supervised and autoencoder baselines with less than 5% of model parameters, achieves 0.6% ~ 1.3% accuracy gain over other tensor-based baselines, and reduces the memory footprint by 9X when compared to standard tensor decomposition algorithms.
MTC: Multiresolution Tensor Completion from Partial and Coarse Observations
Jun 18, 2021
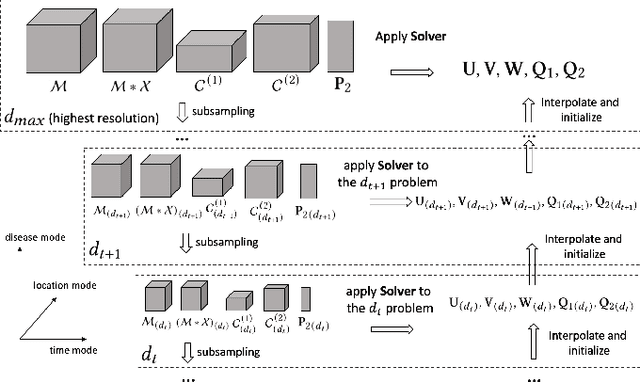
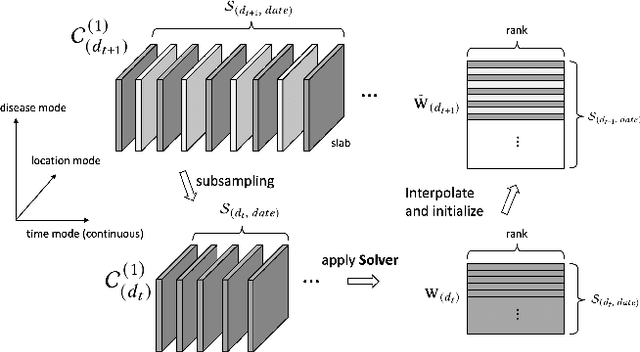

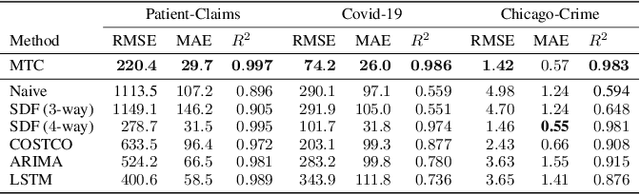
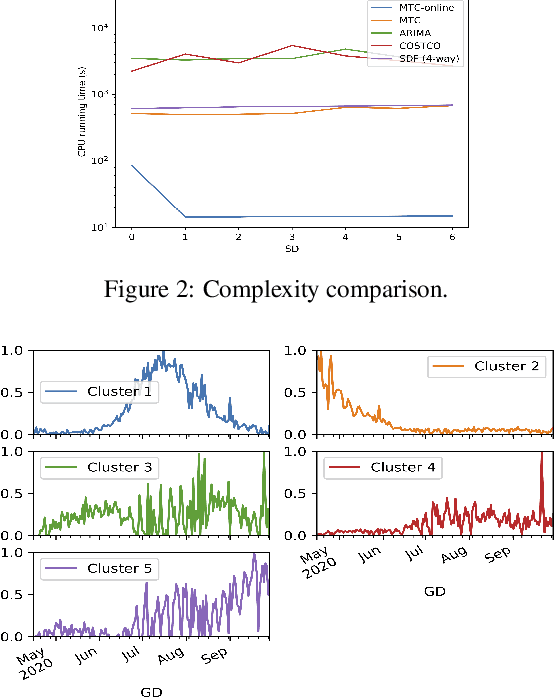
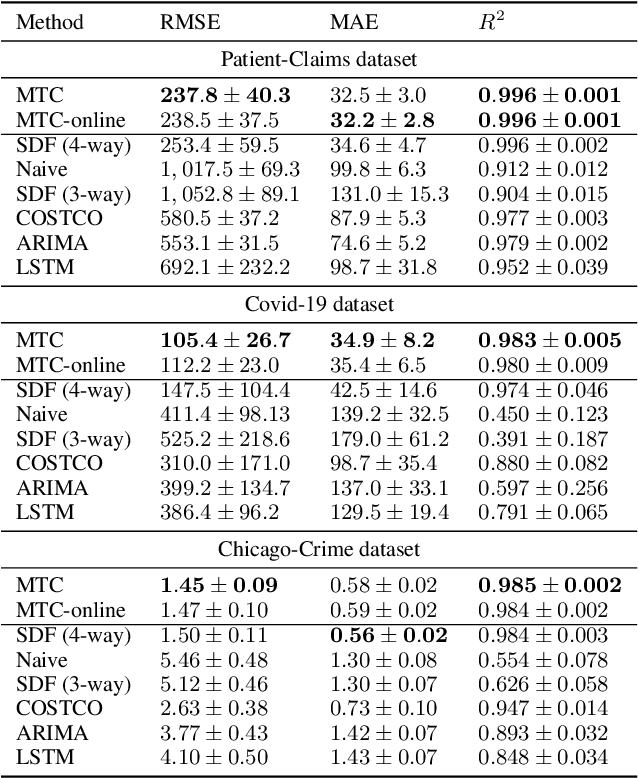
Existing tensor completion formulation mostly relies on partial observations from a single tensor. However, tensors extracted from real-world data are often more complex due to: (i) Partial observation: Only a small subset (e.g., 5%) of tensor elements are available. (ii) Coarse observation: Some tensor modes only present coarse and aggregated patterns (e.g., monthly summary instead of daily reports). In this paper, we are given a subset of the tensor and some aggregated/coarse observations (along one or more modes) and seek to recover the original fine-granular tensor with low-rank factorization. We formulate a coupled tensor completion problem and propose an efficient Multi-resolution Tensor Completion model (MTC) to solve the problem. Our MTC model explores tensor mode properties and leverages the hierarchy of resolutions to recursively initialize an optimization setup, and optimizes on the coupled system using alternating least squares. MTC ensures low computational and space complexity. We evaluate our model on two COVID-19 related spatio-temporal tensors. The experiments show that MTC could provide 65.20% and 75.79% percentage of fitness (PoF) in tensor completion with only 5% fine granular observations, which is 27.96% relative improvement over the best baseline. To evaluate the learned low-rank factors, we also design a tensor prediction task for daily and cumulative disease case predictions, where MTC achieves 50% in PoF and 30% relative improvements over the best baseline.
Multi-version Tensor Completion for Time-delayed Spatio-temporal Data
May 11, 2021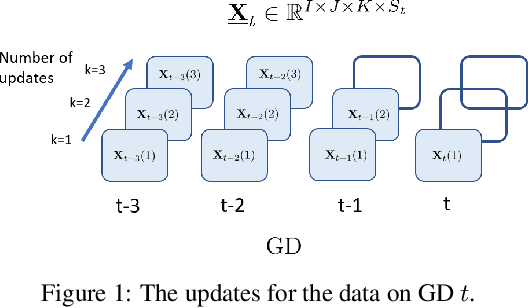
Real-world spatio-temporal data is often incomplete or inaccurate due to various data loading delays. For example, a location-disease-time tensor of case counts can have multiple delayed updates of recent temporal slices for some locations or diseases. Recovering such missing or noisy (under-reported) elements of the input tensor can be viewed as a generalized tensor completion problem. Existing tensor completion methods usually assume that i) missing elements are randomly distributed and ii) noise for each tensor element is i.i.d. zero-mean. Both assumptions can be violated for spatio-temporal tensor data. We often observe multiple versions of the input tensor with different under-reporting noise levels. The amount of noise can be time- or location-dependent as more updates are progressively introduced to the tensor. We model such dynamic data as a multi-version tensor with an extra tensor mode capturing the data updates. We propose a low-rank tensor model to predict the updates over time. We demonstrate that our method can accurately predict the ground-truth values of many real-world tensors. We obtain up to 27.2% lower root mean-squared-error compared to the best baseline method. Finally, we extend our method to track the tensor data over time, leading to significant computational savings.
Change Matters: Medication Change Prediction with Recurrent Residual Networks
May 05, 2021
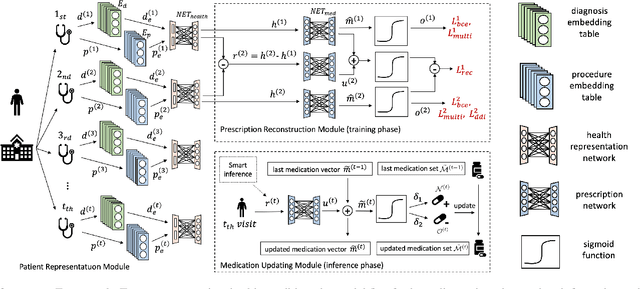
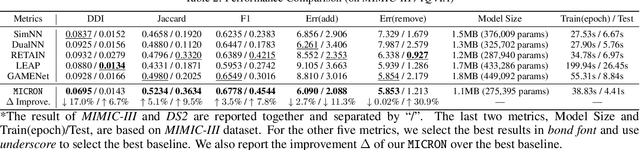
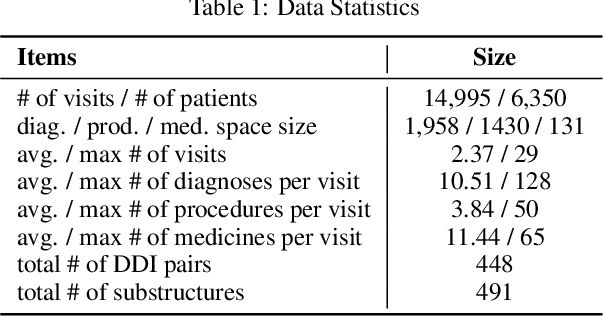
Deep learning is revolutionizing predictive healthcare, including recommending medications to patients with complex health conditions. Existing approaches focus on predicting all medications for the current visit, which often overlaps with medications from previous visits. A more clinically relevant task is to identify medication changes. In this paper, we propose a new recurrent residual network, named MICRON, for medication change prediction. MICRON takes the changes in patient health records as input and learns to update a hidden medication vector and the medication set recurrently with a reconstruction design. The medication vector is like the memory cell that encodes longitudinal information of medications. Unlike traditional methods that require the entire patient history for prediction, MICRON has a residual-based inference that allows for sequential updating based only on new patient features (e.g., new diagnoses in the recent visit) more efficiently. We evaluated MICRON on real inpatient and outpatient datasets. MICRON achieves 3.5% and 7.8% relative improvements over the best baseline in F1 score, respectively. MICRON also requires fewer parameters, which significantly reduces the training time to 38.3s per epoch with 1.5x speed-up.
SafeDrug: Dual Molecular Graph Encoders for Safe Drug Recommendations
May 05, 2021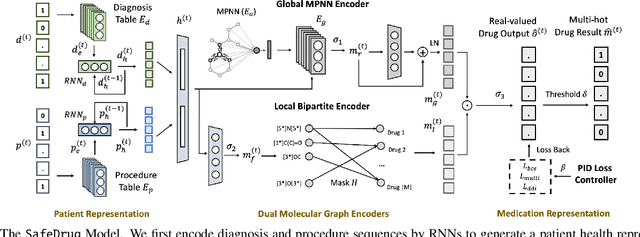
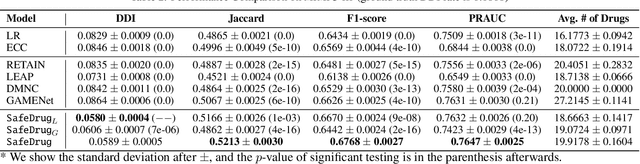
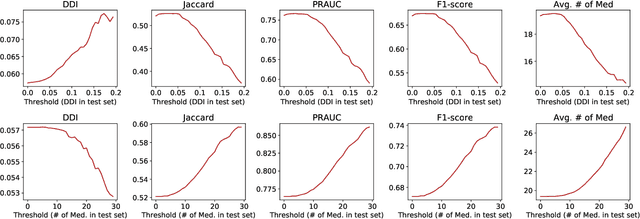
Medication recommendation is an essential task of AI for healthcare. Existing works focused on recommending drug combinations for patients with complex health conditions solely based on their electronic health records. Thus, they have the following limitations: (1) some important data such as drug molecule structures have not been utilized in the recommendation process. (2) drug-drug interactions (DDI) are modeled implicitly, which can lead to sub-optimal results. To address these limitations, we propose a DDI-controllable drug recommendation model named SafeDrug to leverage drugs' molecule structures and model DDIs explicitly. SafeDrug is equipped with a global message passing neural network (MPNN) module and a local bipartite learning module to fully encode the connectivity and functionality of drug molecules. SafeDrug also has a controllable loss function to control DDI levels in the recommended drug combinations effectively. On a benchmark dataset, our SafeDrug is relatively shown to reduce DDI by 19.43% and improves 2.88% on Jaccard similarity between recommended and actually prescribed drug combinations over previous approaches. Moreover, SafeDrug also requires much fewer parameters than previous deep learning-based approaches, leading to faster training by about 14% and around 2x speed-up in inference.
Machine Learning Applications for Therapeutic Tasks with Genomics Data
May 03, 2021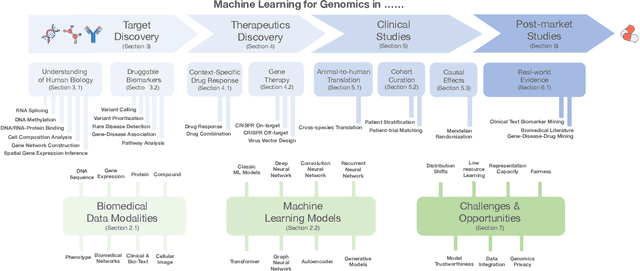
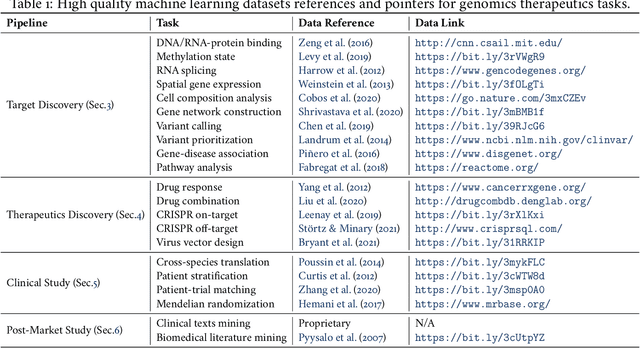
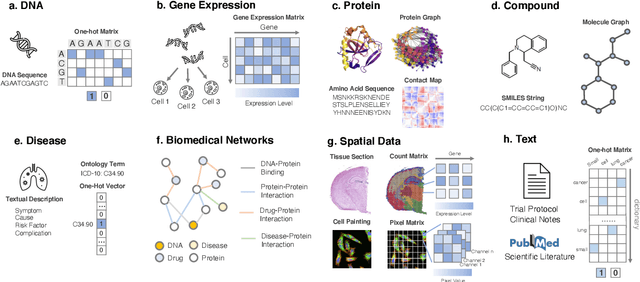
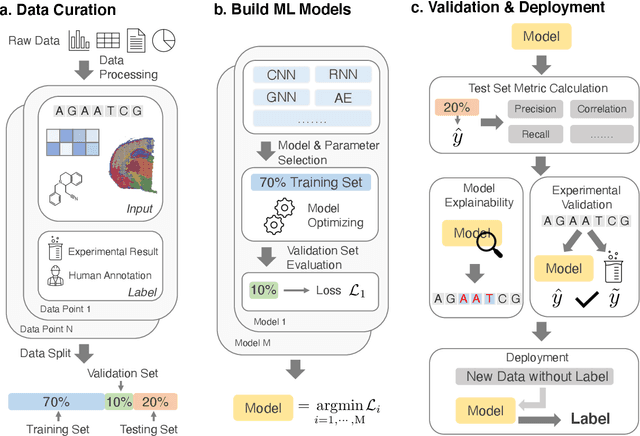
Thanks to the increasing availability of genomics and other biomedical data, many machine learning approaches have been proposed for a wide range of therapeutic discovery and development tasks. In this survey, we review the literature on machine learning applications for genomics through the lens of therapeutic development. We investigate the interplay among genomics, compounds, proteins, electronic health records (EHR), cellular images, and clinical texts. We identify twenty-two machine learning in genomics applications across the entire therapeutics pipeline, from discovering novel targets, personalized medicine, developing gene-editing tools all the way to clinical trials and post-market studies. We also pinpoint seven important challenges in this field with opportunities for expansion and impact. This survey overviews recent research at the intersection of machine learning, genomics, and therapeutic development.
SCRIB: Set-classifier with Class-specific Risk Bounds for Blackbox Models
Mar 05, 2021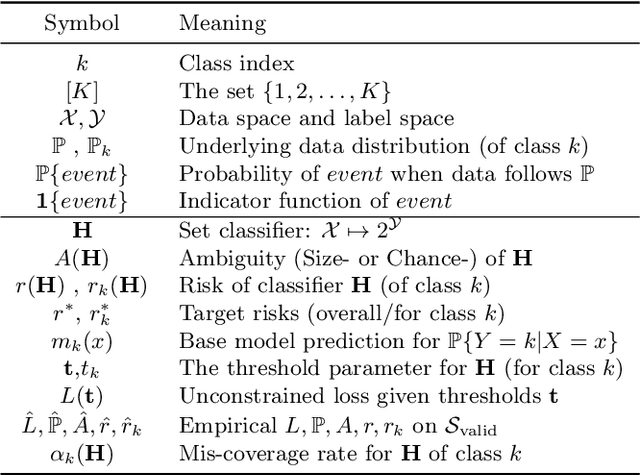
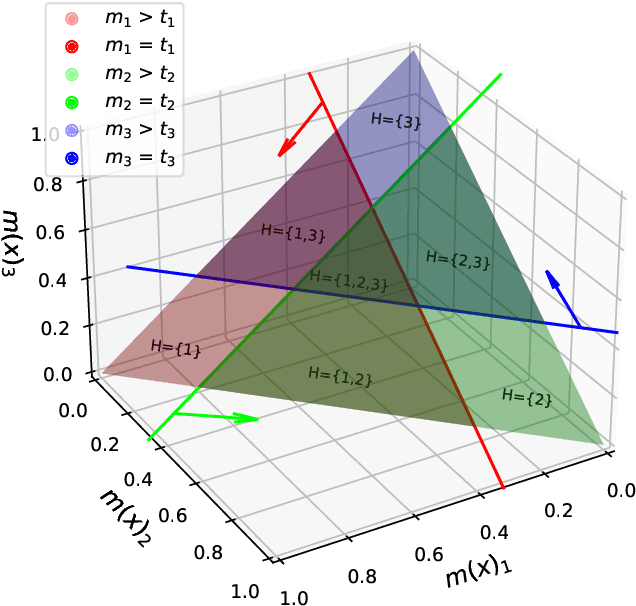
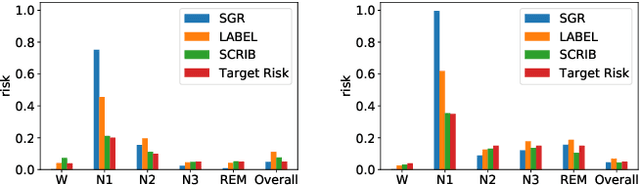
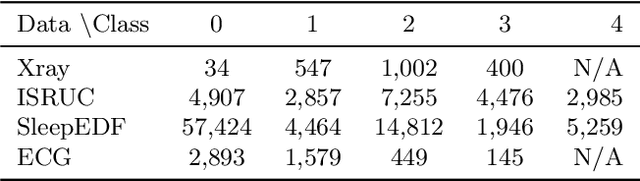
Despite deep learning (DL) success in classification problems, DL classifiers do not provide a sound mechanism to decide when to refrain from predicting. Recent works tried to control the overall prediction risk with classification with rejection options. However, existing works overlook the different significance of different classes. We introduce Set-classifier with Class-specific RIsk Bounds (SCRIB) to tackle this problem, assigning multiple labels to each example. Given the output of a black-box model on the validation set, SCRIB constructs a set-classifier that controls the class-specific prediction risks with a theoretical guarantee. The key idea is to reject when the set classifier returns more than one label. We validated SCRIB on several medical applications, including sleep staging on electroencephalogram (EEG) data, X-ray COVID image classification, and atrial fibrillation detection based on electrocardiogram (ECG) data. SCRIB obtained desirable class-specific risks, which are 35\%-88\% closer to the target risks than baseline methods.
Therapeutics Data Commons: Machine Learning Datasets and Tasks for Therapeutics
Feb 18, 2021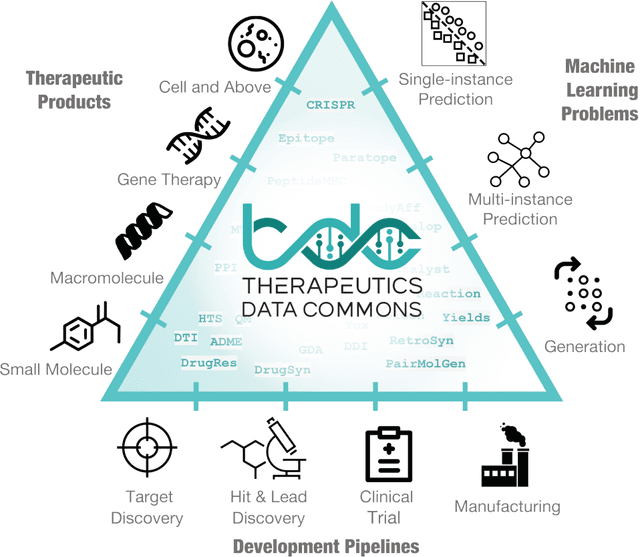
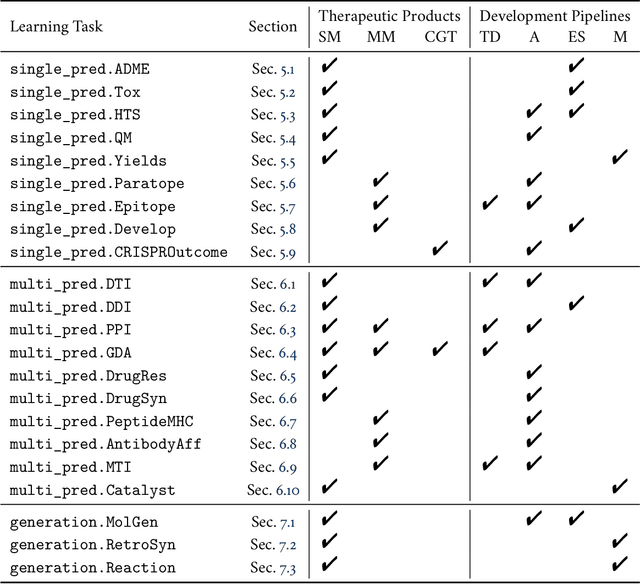
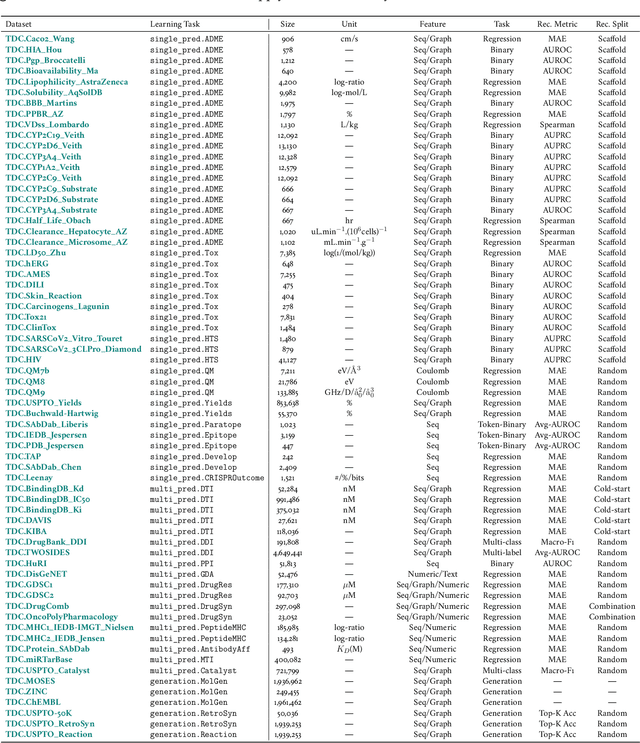
Machine learning for therapeutics is an emerging field with incredible opportunities for innovation and expansion. Despite the initial success, many key challenges remain open. Here, we introduce Therapeutics Data Commons (TDC), the first unifying framework to systematically access and evaluate machine learning across the entire range of therapeutics. At its core, TDC is a collection of curated datasets and learning tasks that can translate algorithmic innovation into biomedical and clinical implementation. To date, TDC includes 66 machine learning-ready datasets from 22 learning tasks, spanning the discovery and development of safe and effective medicines. TDC also provides an ecosystem of tools, libraries, leaderboards, and community resources, including data functions, strategies for systematic model evaluation, meaningful data splits, data processors, and molecule generation oracles. All datasets and learning tasks are integrated and accessible via an open-source library. We envision that TDC can facilitate algorithmic and scientific advances and accelerate development, validation, and transition into production and clinical implementation. TDC is a continuous, open-source initiative, and we invite contributions from the research community. TDC is publicly available at https://tdcommons.ai.