 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edge"cancer detection": models, code, and papers
Double Encoder-Decoder Networks for Gastrointestinal Polyp Segmentation
Oct 05, 2021
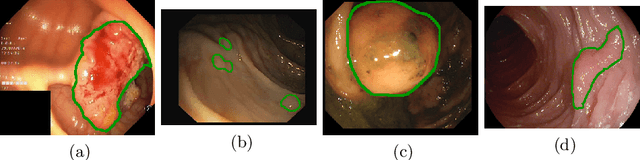

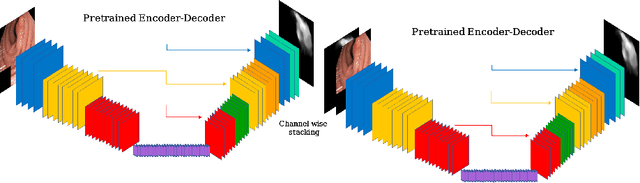
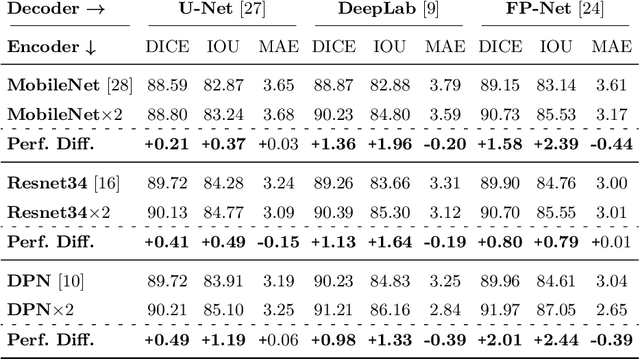
Polyps represent an early sign of the development of Colorectal Cancer. The standard procedure for their detection consists of colonoscopic examination of the gastrointestinal tract. However, the wide range of polyp shapes and visual appearances, as well as the reduced quality of this image modality, turn their automatic identification and segmentation with computational tools into a challenging computer vision task. In this work, we present a new strategy for the delineation of gastrointestinal polyps from endoscopic images based on a direct extension of common encoder-decoder networks for semantic segmentation. In our approach, two pretrained encoder-decoder networks are sequentially stacked: the second network takes as input the concatenation of the original frame and the initial prediction generated by the first network, which acts as an attention mechanism enabling the second network to focus on interesting areas within the image, thereby improving the quality of its predictions. Quantitative evaluation carried out on several polyp segmentation databases shows that double encoder-decoder networks clearly outperform their single encoder-decoder counterparts in all cases. In addition, our best double encoder-decoder combination attains excellent segmentation accuracy and reaches state-of-the-art performance results in all the considered datasets, with a remarkable boost of accuracy on images extracted from datasets not used for training.
Ensemble classifier approach in breast cancer detection and malignancy grading- A review
Apr 11, 2017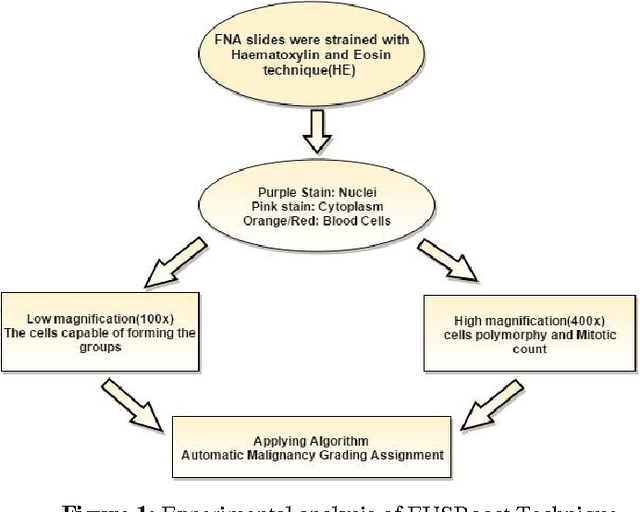



The diagnosed cases of Breast cancer is increasing annually and unfortunately getting converted into a high mortality rate. Cancer, at the early stages, is hard to detect because the malicious cells show similar properties (density) as shown by the non-malicious cells. The mortality ratio could have been minimized if the breast cancer could have been detected in its early stages. But the current systems have not been able to achieve a fully automatic system which is not just capable of detecting the breast cancer but also can detect the stage of it. Estimation of malignancy grading is important in diagnosing the degree of growth of malicious cells as well as in selecting a proper therapy for the patient. Therefore, a complete and efficient clinical decision support system is proposed which is capable of achieving breast cancer malignancy grading scheme very efficiently. The system is based on Image processing and machine learning domains. Classification Imbalance problem, a machine learning problem, occurs when instances of one class is much higher than the instances of the other class resulting in an inefficient classification of samples and hence a bad decision support system. Therefore EUSBoost, ensemble based classifier is proposed which is efficient and is able to outperform other classifiers as it takes the benefits of both-boosting algorithm with Random Undersampling techniques. Also comparison of EUSBoost with other techniques is shown in the paper.
* 10 pages,1 figure,5 tables
Renal Cell Carcinoma Detection and Subtyping with Minimal Point-Based Annotation in Whole-Slide Images
Aug 12, 2020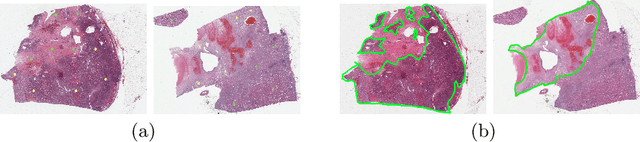
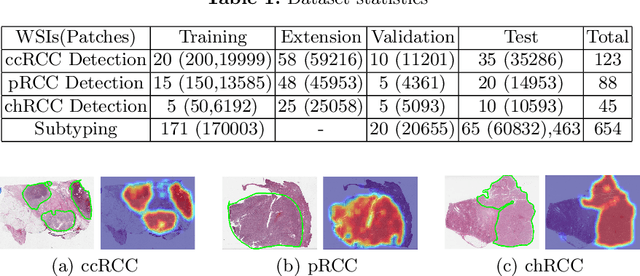
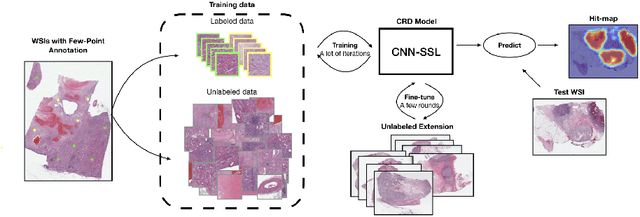
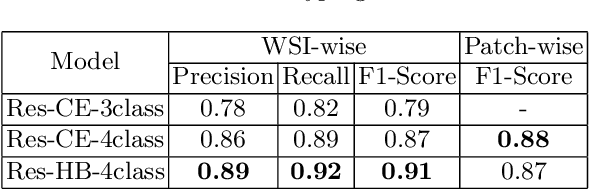
Obtaining a large amount of labeled data in medical imaging is laborious and time-consuming, especially for histopathology. However, it is much easier and cheaper to get unlabeled data from whole-slide images (WSIs). Semi-supervised learning (SSL) is an effective way to utilize unlabeled data and alleviate the need for labeled data. For this reason, we proposed a framework that employs an SSL method to accurately detect cancerous regions with a novel annotation method called Minimal Point-Based annotation, and then utilize the predicted results with an innovative hybrid loss to train a classification model for subtyping. The annotator only needs to mark a few points and label them are cancer or not in each WSI. Experiments on three significant subtypes of renal cell carcinoma (RCC) proved that the performance of the classifier trained with the Min-Point annotated dataset is comparable to a classifier trained with the segmentation annotated dataset for cancer region detection. And the subtyping model outperforms a model trained with only diagnostic labels by 12% in terms of f1-score for testing WSIs.
Delving Deep into Liver Focal Lesion Detection: A Preliminary Study
Jul 24, 2019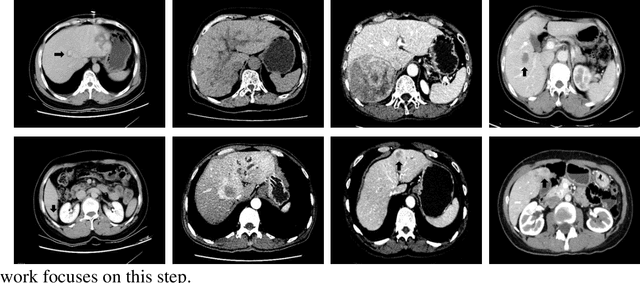
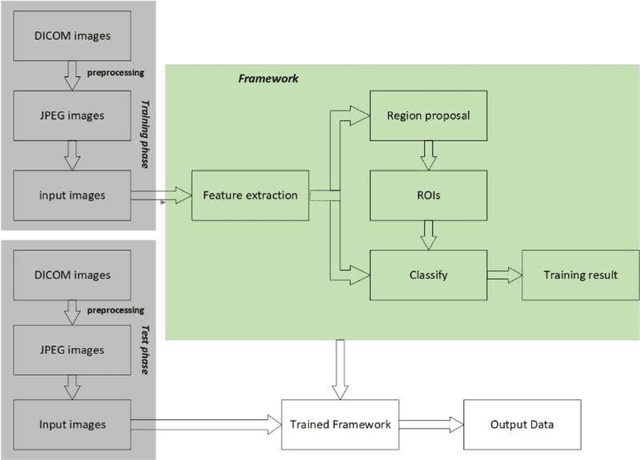

Hepatocellular carcinoma (HCC) is the second most frequent cause of malignancy-related death and is one of the diseases with the highest incidence in the world. Because the liver is the only organ in the human body that is supplied by two major vessels: the hepatic artery and the portal vein, various types of malignant tumors can spread from other organs to the liver. And due to the liver masses' heterogeneous and diffusive shape, the tumor lesions are very difficult to be recognized, thus automatic lesion detection is necessary for the doctors with huge workloads. To assist doctors, this work uses the existing large-scale annotation medical image data to delve deep into liver lesion detection from multiple directions. To solve technical difficulties, such as the image-recognition task, traditional deep learning with convolution neural networks (CNNs) has been widely applied in recent years. However, this kind of neural network, such as Faster Regions with CNN features (R-CNN), cannot leverage the spatial information because it is applied in natural images (2D) rather than medical images (3D), such as computed tomography (CT) images. To address this issue, we propose a novel algorithm that is appropriate for liver CT imaging. Furthermore, according to radiologists' experience in clinical diagnosis and the characteristics of CT images of liver cancer, a liver cancer-detection framework with CNN, including image processing, feature extraction, region proposal, image registration, and classification recognition, was proposed to facilitate the effective detection of liver lesions.
Machine Learning Characterization of Cancer Patients-Derived Extracellular Vesicles using Vibrational Spectroscopies
Aug 04, 2021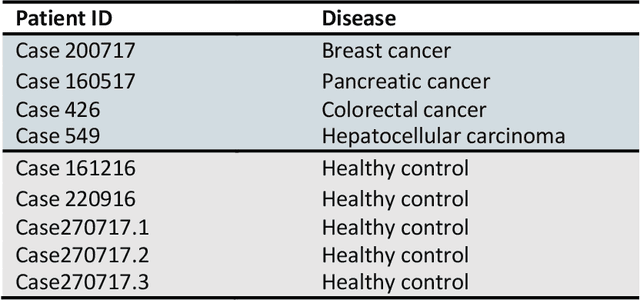
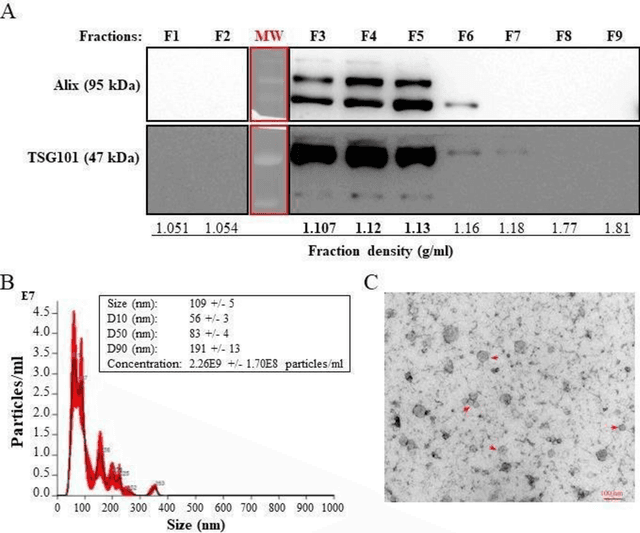
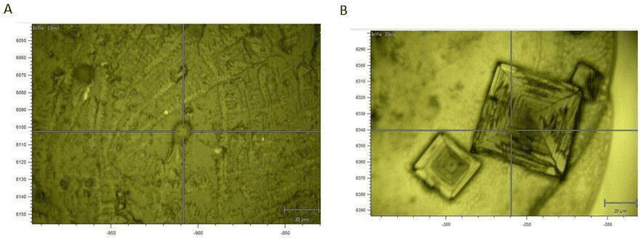
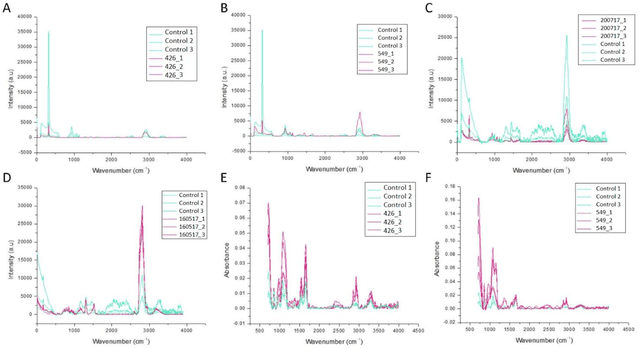
The early detection of cancer is a challenging problem in medicine. The blood sera of cancer patients are enriched with heterogeneous secretory lipid bound extracellular vesicles (EVs), which present a complex repertoire of information and biomarkers, representing their cell of origin, that are being currently studied in the field of liquid biopsy and cancer screening. Vibrational spectroscopies provide non-invasive approaches for the assessment of structural and biophysical properties in complex biological samples. In this study, multiple Raman spectroscopy measurements were performed on the EVs extracted from the blood sera of 9 patients consisting of four different cancer subtypes (colorectal cancer, hepatocellular carcinoma, breast cancer and pancreatic cancer) and five healthy patients (controls). FTIR(Fourier Transform Infrared) spectroscopy measurements were performed as a complementary approach to Raman analysis, on two of the four cancer subtypes. The AdaBoost Random Forest Classifier, Decision Trees, and Support Vector Machines (SVM) distinguished the baseline corrected Raman spectra of cancer EVs from those of healthy controls (18 spectra) with a classification accuracy of greater than 90% when reduced to a spectral frequency range of 1800 to 1940 inverse cm, and subjected to a 0.5 training/testing split. FTIR classification accuracy on 14 spectra showed an 80% classification accuracy. Our findings demonstrate that basic machine learning algorithms are powerful tools to distinguish the complex vibrational spectra of cancer patient EVs from those of healthy patients. These experimental methods hold promise as valid and efficient liquid biopsy for machine intelligence-assisted early cancer screening.
Preserving Dense Features for Ki67 Nuclei Detection
Nov 10, 2021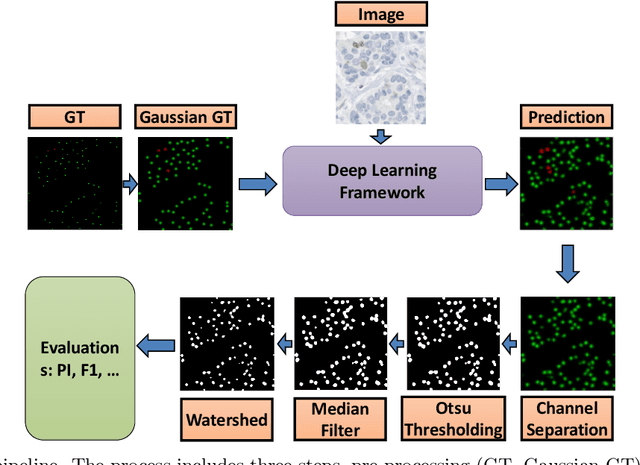
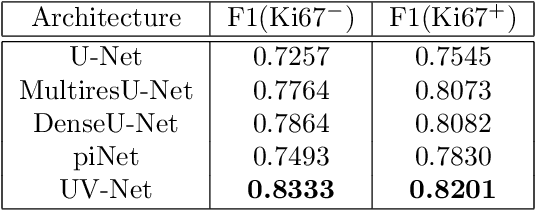
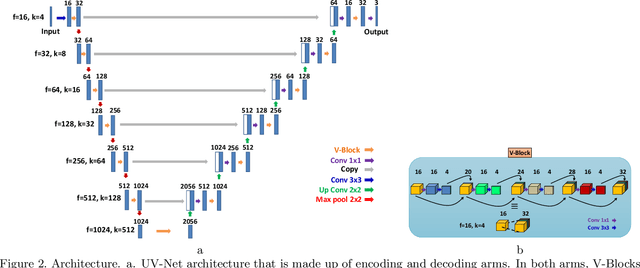
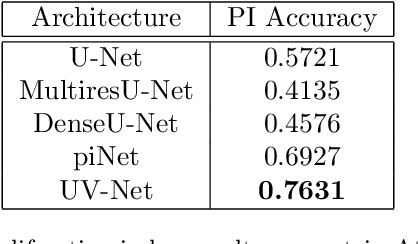
Nuclei detection is a key task in Ki67 proliferation index estimation in breast cancer images. Deep learning algorithms have shown strong potential in nuclei detection tasks. However, they face challenges when applied to pathology images with dense medium and overlapping nuclei since fine details are often diluted or completely lost by early maxpooling layers. This paper introduces an optimized UV-Net architecture, specifically developed to recover nuclear details with high-resolution through feature preservation for Ki67 proliferation index computation. UV-Net achieves an average F1-score of 0.83 on held-out test patch data, while other architectures obtain 0.74-0.79. On tissue microarrays (unseen) test data obtained from multiple centers, UV-Net's accuracy exceeds other architectures by a wide margin, including 9-42\% on Ontario Veterinary College, 7-35\% on Protein Atlas and 0.3-3\% on University Health Network.
Automated Detection of Acute Leukemia using K-mean Clustering Algorithm
Mar 06, 2018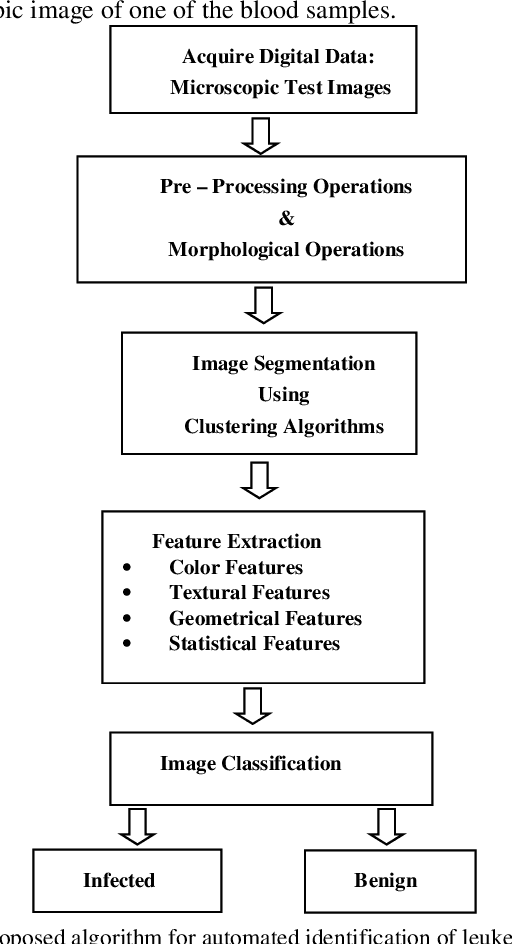
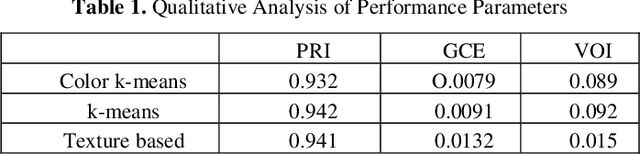
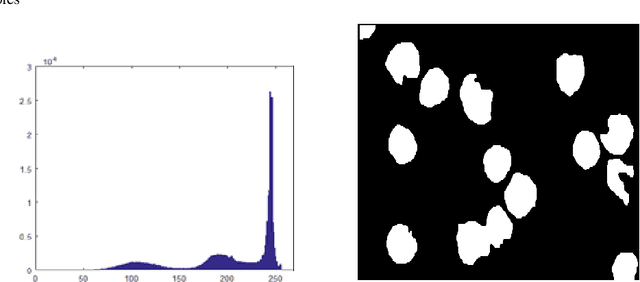
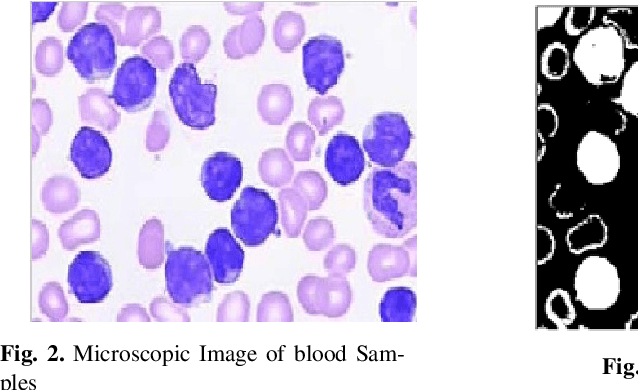
Leukemia is a hematologic cancer which develops in blood tissue and triggers rapid production of immature and abnormal shaped white blood cells. Based on statistics it is found that the leukemia is one of the leading causes of death in men and women alike. Microscopic examination of blood sample or bone marrow smear is the most effective technique for diagnosis of leukemia. Pathologists analyze microscopic samples to make diagnostic assessments on the basis of characteristic cell features. Recently, computerized methods for cancer detection have been explored towards minimizing human intervention and providing accurate clinical information. This paper presents an algorithm for automated image based acute leukemia detection systems. The method implemented uses basic enhancement, morphology, filtering and segmenting technique to extract region of interest using k-means clustering algorithm. The proposed algorithm achieved an accuracy of 92.8% and is tested with Nearest Neighbor (KNN) and Naive Bayes Classifier on the data-set of 60 samples.
* Presented in ICCCCS 2016
Assessing domain adaptation techniques for mitosis detection in multi-scanner breast cancer histopathology images
Sep 25, 2021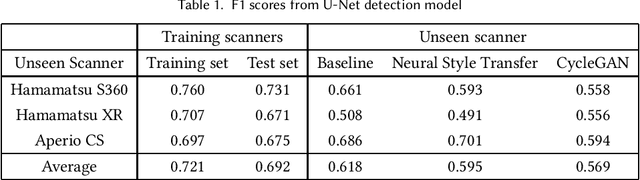
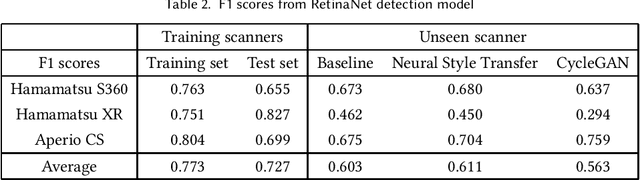
Breast cancer is the most prevalent cancer worldwide and is increasing in incidence, with over two million new cases now diagnosed each year. As part of diagnostic tumour grading, histopathologists manually count the number of dividing cells (mitotic figures) in a sample. Since the process is subjective and time-consuming, artificial intelligence (AI) methods have been developed to automate the process, however these methods often perform poorly when applied to data from outside of the original (training) domain, i.e. they do not generalise well to variations in histological background, staining protocols, or scanner types. Style transfer, a form of domain adaptation, provides the means to transform images from different domains to a shared visual appearance and have been adopted in various applications to mitigate the issue of domain shift. In this paper we train two mitosis detection models and two style transfer methods and evaluate the usefulness of the latter for improving mitosis detection performance in images digitised using different scanners. We found that the best of these models, U-Net without style transfer, achieved an F1-score of 0.693 on the MIDOG 2021 preliminary test set.
Transport-based analysis, modeling, and learning from signal and data distributions
Sep 15, 2016
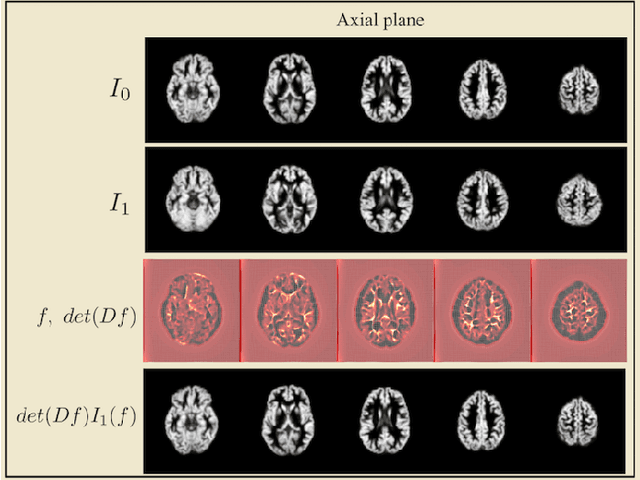
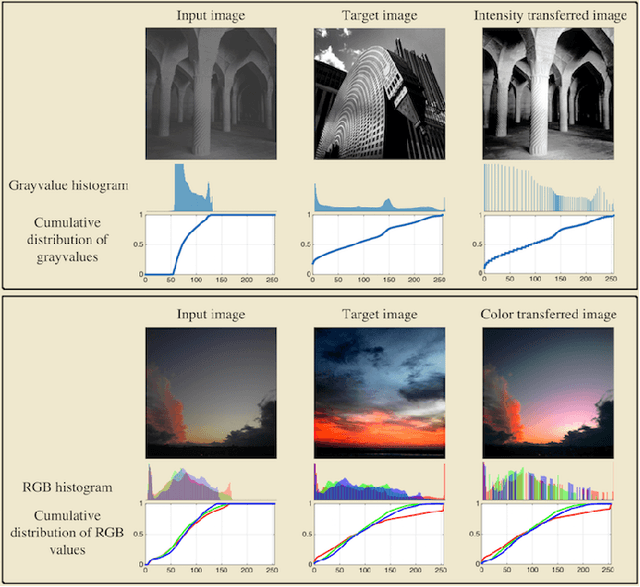
Transport-based techniques for signal and data analysis have received increased attention recently. Given their abilities to provide accurate generative models for signal intensities and other data distributions, they have been used in a variety of applications including content-based retrieval, cancer detection, image super-resolution, and statistical machine learning, to name a few, and shown to produce state of the art in several applications. Moreover, the geometric characteristics of transport-related metrics have inspired new kinds of algorithms for interpreting the meaning of data distributions. Here we provide an overview of the mathematical underpinnings of mass transport-related methods, including numerical implementation, as well as a review, with demonstrations, of several applications.