 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgecancer detection
Cancer detection using Artificial Intelligence (AI) involves leveraging advanced machine learning algorithms and techniques to identify and diagnose cancer from various medical data sources. The goal is to enhance early detection, improve diagnostic accuracy, and potentially reduce the need for invasive procedures.
Papers and Code
Model-Independent Machine Learning Approach for Nanometric Axial Localization and Tracking
May 20, 2025Accurately tracking particles and determining their position along the optical axis is a major challenge in optical microscopy, especially when extremely high precision is needed. In this study, we introduce a deep learning approach using convolutional neural networks (CNNs) that can determine axial positions from dual-focal plane images without relying on predefined models. Our method achieves an axial localization accuracy of 40 nanometers - six times better than traditional single-focal plane techniques. The model's simple design and strong performance make it suitable for a wide range of uses, including dark matter detection, proton therapy for cancer, and radiation protection in space. It also shows promise in fields like biological imaging, materials science, and environmental monitoring. This work highlights how machine learning can turn complex image data into reliable, precise information, offering a flexible and powerful tool for many scientific applications.
Discovering Pathology Rationale and Token Allocation for Efficient Multimodal Pathology Reasoning
May 21, 2025Multimodal pathological image understanding has garnered widespread interest due to its potential to improve diagnostic accuracy and enable personalized treatment through integrated visual and textual data. However, existing methods exhibit limited reasoning capabilities, which hamper their ability to handle complex diagnostic scenarios. Additionally, the enormous size of pathological images leads to severe computational burdens, further restricting their practical deployment. To address these limitations, we introduce a novel bilateral reinforcement learning framework comprising two synergistic branches. One reinforcement branch enhances the reasoning capability by enabling the model to learn task-specific decision processes, i.e., pathology rationales, directly from labels without explicit reasoning supervision. While the other branch dynamically allocates a tailored number of tokens to different images based on both their visual content and task context, thereby optimizing computational efficiency. We apply our method to various pathological tasks such as visual question answering, cancer subtyping, and lesion detection. Extensive experiments show an average +41.7 absolute performance improvement with 70.3% lower inference costs over the base models, achieving both reasoning accuracy and computational efficiency.
A Comprehensive Study on Medical Image Segmentation using Deep Neural Networks
Jun 04, 2025Over the past decade, Medical Image Segmentation (MIS) using Deep Neural Networks (DNNs) has achieved significant performance improvements and holds great promise for future developments. This paper presents a comprehensive study on MIS based on DNNs. Intelligent Vision Systems are often evaluated based on their output levels, such as Data, Information, Knowledge, Intelligence, and Wisdom (DIKIW),and the state-of-the-art solutions in MIS at these levels are the focus of research. Additionally, Explainable Artificial Intelligence (XAI) has become an important research direction, as it aims to uncover the "black box" nature of previous DNN architectures to meet the requirements of transparency and ethics. The study emphasizes the importance of MIS in disease diagnosis and early detection, particularly for increasing the survival rate of cancer patients through timely diagnosis. XAI and early prediction are considered two important steps in the journey from "intelligence" to "wisdom." Additionally, the paper addresses existing challenges and proposes potential solutions to enhance the efficiency of implementing DNN-based MIS.
Lung Nodule-SSM: Self-Supervised Lung Nodule Detection and Classification in Thoracic CT Images
May 21, 2025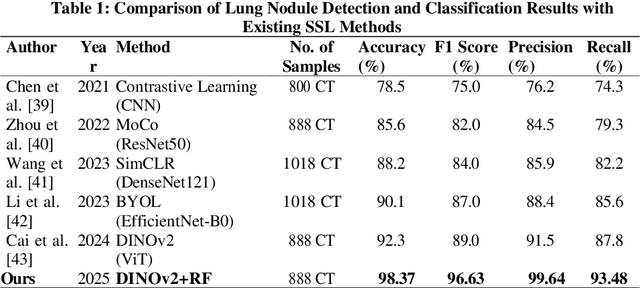
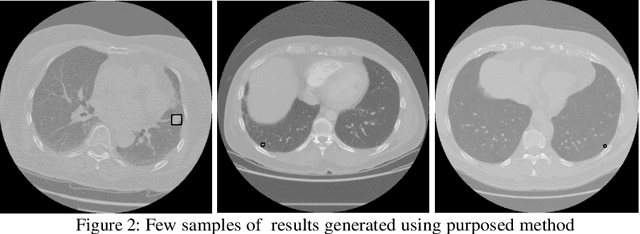
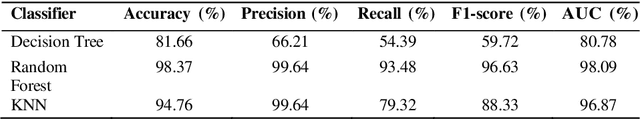
Lung cancer remains among the deadliest types of cancer in recent decades, and early lung nodule detection is crucial for improving patient outcomes. The limited availability of annotated medical imaging data remains a bottleneck in developing accurate computer-aided diagnosis (CAD) systems. Self-supervised learning can help leverage large amounts of unlabeled data to develop more robust CAD systems. With the recent advent of transformer-based architecture and their ability to generalize to unseen tasks, there has been an effort within the healthcare community to adapt them to various medical downstream tasks. Thus, we propose a novel "LungNodule-SSM" method, which utilizes selfsupervised learning with DINOv2 as a backbone to enhance lung nodule detection and classification without annotated data. Our methodology has two stages: firstly, the DINOv2 model is pre-trained on unlabeled CT scans to learn robust feature representations, then secondly, these features are fine-tuned using transformer-based architectures for lesionlevel detection and accurate lung nodule diagnosis. The proposed method has been evaluated on the challenging LUNA 16 dataset, consisting of 888 CT scans, and compared with SOTA methods. Our experimental results show the superiority of our proposed method with an accuracy of 98.37%, explaining its effectiveness in lung nodule detection. The source code, datasets, and pre-processed data can be accessed using the link:https://github.com/EMeRALDsNRPU/Lung-Nodule-SSM-Self-Supervised-Lung-Nodule-Detection-and-Classification/tree/main
Deep Learning Enabled Segmentation, Classification and Risk Assessment of Cervical Cancer
May 21, 2025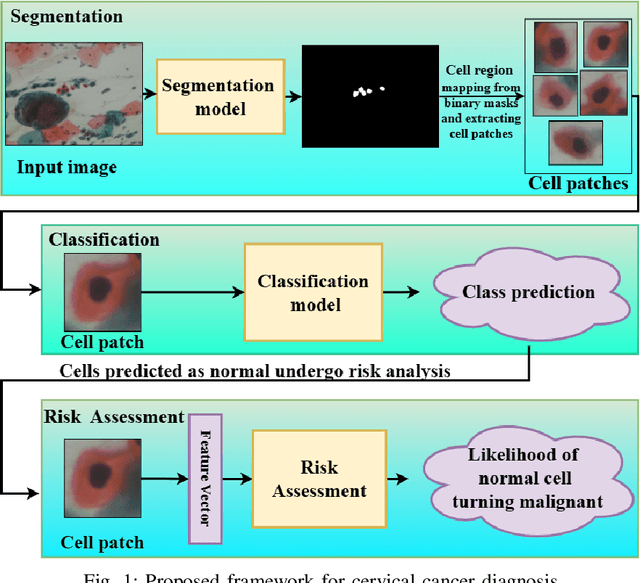
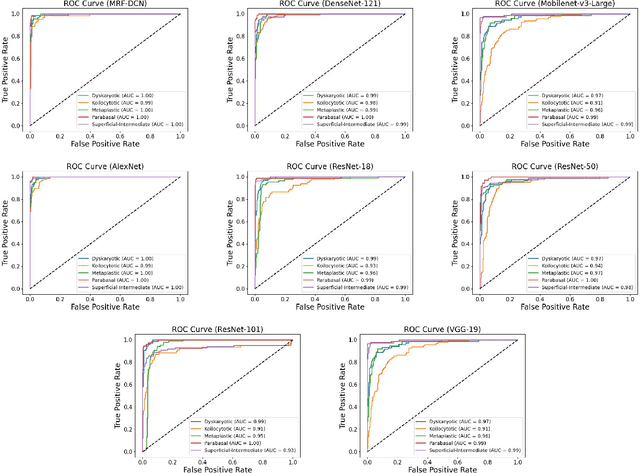
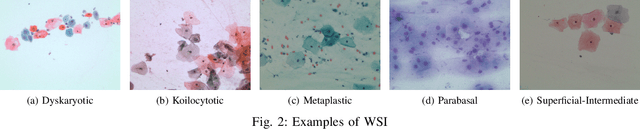
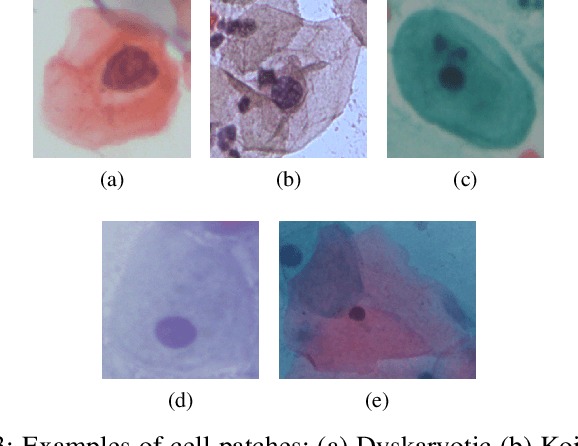
Cervical cancer, the fourth leading cause of cancer in women globally, requires early detection through Pap smear tests to identify precancerous changes and prevent disease progression. In this study, we performed a focused analysis by segmenting the cellular boundaries and drawing bounding boxes to isolate the cancer cells. A novel Deep Learning (DL) architecture, the ``Multi-Resolution Fusion Deep Convolutional Network", was proposed to effectively handle images with varying resolutions and aspect ratios, with its efficacy showcased using the SIPaKMeD dataset. The performance of this DL model was observed to be similar to the state-of-the-art models, with accuracy variations of a mere 2\% to 3\%, achieved using just 1.7 million learnable parameters, which is approximately 85 times less than the VGG-19 model. Furthermore, we introduced a multi-task learning technique that simultaneously performs segmentation and classification tasks and begets an Intersection over Union score of 0.83 and a classification accuracy of 90\%. The final stage of the workflow employs a probabilistic approach for risk assessment, extracting feature vectors to predict the likelihood of normal cells progressing to malignant states, which can be utilized for the prognosis of cervical cancer.
Towards Facilitated Fairness Assessment of AI-based Skin Lesion Classifiers Through GenAI-based Image Synthesis
Jul 23, 2025Recent advancements in Deep Learning and its application on the edge hold great potential for the revolution of routine screenings for skin cancers like Melanoma. Along with the anticipated benefits of this technology, potential dangers arise from unforseen and inherent biases. Thus, assessing and improving the fairness of such systems is of utmost importance. A key challenge in fairness assessment is to ensure that the evaluation dataset is sufficiently representative of different Personal Identifiable Information (PII) (sex, age, and race) and other minority groups. Against the backdrop of this challenge, this study leverages the state-of-the-art Generative AI (GenAI) LightningDiT model to assess the fairness of publicly available melanoma classifiers. The results suggest that fairness assessment using highly realistic synthetic data is a promising direction. Yet, our findings indicate that verifying fairness becomes difficult when the melanoma-detection model used for evaluation is trained on data that differ from the dataset underpinning the synthetic images. Nonetheless, we propose that our approach offers a valuable new avenue for employing synthetic data to gauge and enhance fairness in medical-imaging GenAI systems.
Learning from Anatomy: Supervised Anatomical Pretraining (SAP) for Improved Metastatic Bone Disease Segmentation in Whole-Body MRI
Jun 24, 2025The segmentation of metastatic bone disease (MBD) in whole-body MRI (WB-MRI) is a challenging problem. Due to varying appearances and anatomical locations of lesions, ambiguous boundaries, and severe class imbalance, obtaining reliable segmentations requires large, well-annotated datasets capturing lesion variability. Generating such datasets requires substantial time and expertise, and is prone to error. While self-supervised learning (SSL) can leverage large unlabeled datasets, learned generic representations often fail to capture the nuanced features needed for accurate lesion detection. In this work, we propose a Supervised Anatomical Pretraining (SAP) method that learns from a limited dataset of anatomical labels. First, an MRI-based skeletal segmentation model is developed and trained on WB-MRI scans from healthy individuals for high-quality skeletal delineation. Then, we compare its downstream efficacy in segmenting MBD on a cohort of 44 patients with metastatic prostate cancer, against both a baseline random initialization and a state-of-the-art SSL method. SAP significantly outperforms both the baseline and SSL-pretrained models, achieving a normalized surface Dice of 0.76 and a Dice coefficient of 0.64. The method achieved a lesion detection F2 score of 0.44, improving on 0.24 (baseline) and 0.31 (SSL). When considering only clinically relevant lesions larger than 1~ml, SAP achieves a detection sensitivity of 100% in 28 out of 32 patients. Learning bone morphology from anatomy yields an effective and domain-relevant inductive bias that can be leveraged for the downstream segmentation task of bone lesions. All code and models are made publicly available.
Anomaly Detection and Improvement of Clusters using Enhanced K-Means Algorithm
May 30, 2025This paper introduces a unified approach to cluster refinement and anomaly detection in datasets. We propose a novel algorithm that iteratively reduces the intra-cluster variance of N clusters until a global minimum is reached, yielding tighter clusters than the standard k-means algorithm. We evaluate the method using intrinsic measures for unsupervised learning, including the silhouette coefficient, Calinski-Harabasz index, and Davies-Bouldin index, and extend it to anomaly detection by identifying points whose assignment causes a significant variance increase. External validation on synthetic data and the UCI Breast Cancer and UCI Wine Quality datasets employs the Jaccard similarity score, V-measure, and F1 score. Results show variance reductions of 18.7% and 88.1% on the synthetic and Wine Quality datasets, respectively, along with accuracy and F1 score improvements of 22.5% and 20.8% on the Wine Quality dataset.
Improving Oral Cancer Outcomes Through Machine Learning and Dimensionality Reduction
Jun 11, 2025Oral cancer presents a formidable challenge in oncology, necessitating early diagnosis and accurate prognosis to enhance patient survival rates. Recent advancements in machine learning and data mining have revolutionized traditional diagnostic methodologies, providing sophisticated and automated tools for differentiating between benign and malignant oral lesions. This study presents a comprehensive review of cutting-edge data mining methodologies, including Neural Networks, K-Nearest Neighbors (KNN), Support Vector Machines (SVM), and ensemble learning techniques, specifically applied to the diagnosis and prognosis of oral cancer. Through a rigorous comparative analysis, our findings reveal that Neural Networks surpass other models, achieving an impressive classification accuracy of 93,6 % in predicting oral cancer. Furthermore, we underscore the potential benefits of integrating feature selection and dimensionality reduction techniques to enhance model performance. These insights underscore the significant promise of advanced data mining techniques in bolstering early detection, optimizing treatment strategies, and ultimately improving patient outcomes in the realm of oral oncology.
A Narrative Review on Large AI Models in Lung Cancer Screening, Diagnosis, and Treatment Planning
Jun 08, 2025Lung cancer remains one of the most prevalent and fatal diseases worldwide, demanding accurate and timely diagnosis and treatment. Recent advancements in large AI models have significantly enhanced medical image understanding and clinical decision-making. This review systematically surveys the state-of-the-art in applying large AI models to lung cancer screening, diagnosis, prognosis, and treatment. We categorize existing models into modality-specific encoders, encoder-decoder frameworks, and joint encoder architectures, highlighting key examples such as CLIP, BLIP, Flamingo, BioViL-T, and GLoRIA. We further examine their performance in multimodal learning tasks using benchmark datasets like LIDC-IDRI, NLST, and MIMIC-CXR. Applications span pulmonary nodule detection, gene mutation prediction, multi-omics integration, and personalized treatment planning, with emerging evidence of clinical deployment and validation. Finally, we discuss current limitations in generalizability, interpretability, and regulatory compliance, proposing future directions for building scalable, explainable, and clinically integrated AI systems. Our review underscores the transformative potential of large AI models to personalize and optimize lung cancer care.