 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to Edgeh-MINT: Modeling Pocket-Ligand Binding with Hierarchical Molecular Interaction Network
Apr 25, 2026Accurate molecular representations are critical for drug discovery, and a central challenge lies in capturing the chemical environment of molecular fragments, as key interactions, such as H-bond and π stacking, occur only under specific local conditions. Most existing approaches represent molecules as atom-level graphs; however, atom-level representations can hardly express higher-order chemical context (e.g., stereochemistry, lone pairs, conjugation). Fragment-based methods (e.g., principal subgraph, predefined functional groups) fail to preserve essential information such as chirality, aromaticity, and ionic states. This work addresses these limitations from two aspects. (i) OverlapBPE tokenization. We propose a novel data-driven molecule tokenization method. Unlike existing approaches, our method allows overlapping fragments, reflecting the inherently fuzzy boundaries of small-molecule substructures and, together with enriched chemical information at the token level, thereby preserving a more complete chemical context. (ii) h-MINT model. OverlapBPE induces many-to-many atom-fragment mappings, which necessitate a new hierarchical architecture. We therefore develop a hierarchical molecular interaction network capable of jointly modeling interactions at both atom and fragment levels. By supporting fragment overlaps, the model naturally accommodates the many-to-many atom-fragment mappings introduced by the OverlapBPE scheme. Extensive evaluation against state-of-the-art methods shows our method improves binding affinity prediction by 2-4% Pearson/Spearman correlation on PDBBind and LBA, enhances virtual screening by 1-3% in key metrics on DUD-E and LIT-PCBA, and achieves the best overall HTS performance on PubChem assays. Further analysis demonstrates that our method effectively captures interactive information while maintaining good generalization.
Meta Flow Matching: Integrating Vector Fields on the Wasserstein Manifold
Aug 26, 2024



Numerous biological and physical processes can be modeled as systems of interacting entities evolving continuously over time, e.g. the dynamics of communicating cells or physical particles. Learning the dynamics of such systems is essential for predicting the temporal evolution of populations across novel samples and unseen environments. Flow-based models allow for learning these dynamics at the population level - they model the evolution of the entire distribution of samples. However, current flow-based models are limited to a single initial population and a set of predefined conditions which describe different dynamics. We argue that multiple processes in natural sciences have to be represented as vector fields on the Wasserstein manifold of probability densities. That is, the change of the population at any moment in time depends on the population itself due to the interactions between samples. In particular, this is crucial for personalized medicine where the development of diseases and their respective treatment response depends on the microenvironment of cells specific to each patient. We propose Meta Flow Matching (MFM), a practical approach to integrating along these vector fields on the Wasserstein manifold by amortizing the flow model over the initial populations. Namely, we embed the population of samples using a Graph Neural Network (GNN) and use these embeddings to train a Flow Matching model. This gives MFM the ability to generalize over the initial distributions unlike previously proposed methods. We demonstrate the ability of MFM to improve prediction of individual treatment responses on a large scale multi-patient single-cell drug screen dataset.
Sparsity regularization via tree-structured environments for disentangled representations
Jun 10, 2024



Many causal systems such as biological processes in cells can only be observed indirectly via measurements, such as gene expression. Causal representation learning -- the task of correctly mapping low-level observations to latent causal variables -- could advance scientific understanding by enabling inference of latent variables such as pathway activation. In this paper, we develop methods for inferring latent variables from multiple related datasets (environments) and tasks. As a running example, we consider the task of predicting a phenotype from gene expression, where we often collect data from multiple cell types or organisms that are related in known ways. The key insight is that the mapping from latent variables driven by gene expression to the phenotype of interest changes sparsely across closely related environments. To model sparse changes, we introduce Tree-Based Regularization (TBR), an objective that minimizes both prediction error and regularizes closely related environments to learn similar predictors. We prove that under assumptions about the degree of sparse changes, TBR identifies the true latent variables up to some simple transformations. We evaluate the theory empirically with both simulations and ground-truth gene expression data. We find that TBR recovers the latent causal variables better than related methods across these settings, even under settings that violate some assumptions of the theory.
Leveraging Structure Between Environments: Phylogenetic Regularization Incentivizes Disentangled Representations
May 30, 2024Many causal systems such as biological processes in cells can only be observed indirectly via measurements, such as gene expression. Causal representation learning -- the task of correctly mapping low-level observations to latent causal variables -- could advance scientific understanding by enabling inference of latent variables such as pathway activation. In this paper, we develop methods for inferring latent variables from multiple related datasets (environments) and tasks. As a running example, we consider the task of predicting a phenotype from gene expression, where we often collect data from multiple cell types or organisms that are related in known ways. The key insight is that the mapping from latent variables driven by gene expression to the phenotype of interest changes sparsely across closely related environments. To model sparse changes, we introduce Tree-Based Regularization (TBR), an objective that minimizes both prediction error and regularizes closely related environments to learn similar predictors. We prove that under assumptions about the degree of sparse changes, TBR identifies the true latent variables up to some simple transformations. We evaluate the theory empirically with both simulations and ground-truth gene expression data. We find that TBR recovers the latent causal variables better than related methods across these settings, even under settings that violate some assumptions of the theory.
PhyloGFN: Phylogenetic inference with generative flow networks
Oct 12, 2023Phylogenetics is a branch of computational biology that studies the evolutionary relationships among biological entities. Its long history and numerous applications notwithstanding, inference of phylogenetic trees from sequence data remains challenging: the high complexity of tree space poses a significant obstacle for the current combinatorial and probabilistic techniques. In this paper, we adopt the framework of generative flow networks (GFlowNets) to tackle two core problems in phylogenetics: parsimony-based and Bayesian phylogenetic inference. Because GFlowNets are well-suited for sampling complex combinatorial structures, they are a natural choice for exploring and sampling from the multimodal posterior distribution over tree topologies and evolutionary distances. We demonstrate that our amortized posterior sampler, PhyloGFN, produces diverse and high-quality evolutionary hypotheses on real benchmark datasets. PhyloGFN is competitive with prior works in marginal likelihood estimation and achieves a closer fit to the target distribution than state-of-the-art variational inference methods.
Neural representation and generation for RNA secondary structures
Feb 01, 2021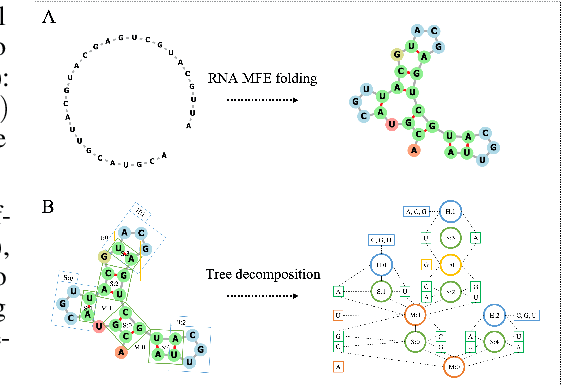
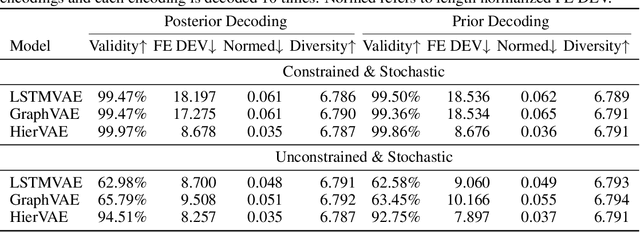
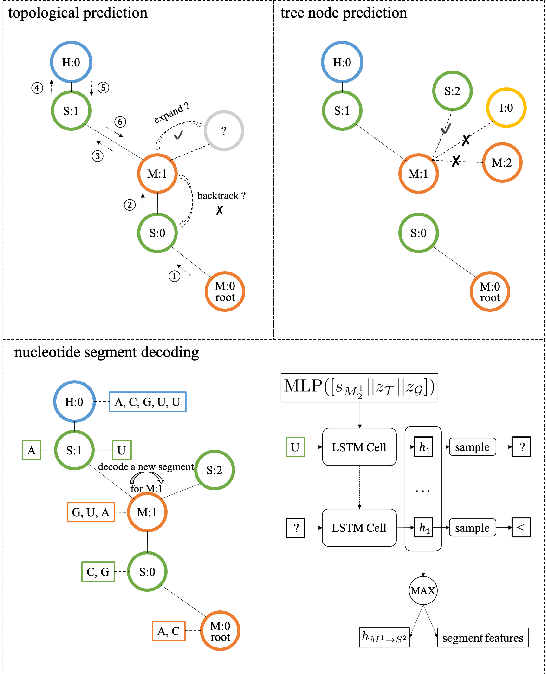
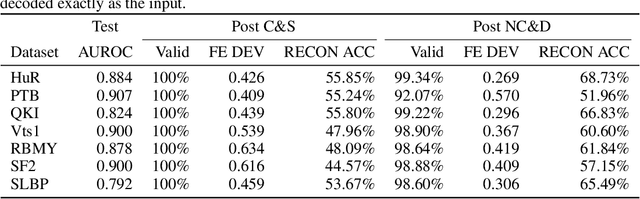
Our work is concerned with the generation and targeted design of RNA, a type of genetic macromolecule that can adopt complex structures which influence their cellular activities and functions. The design of large scale and complex biological structures spurs dedicated graph-based deep generative modeling techniques, which represents a key but underappreciated aspect of computational drug discovery. In this work, we investigate the principles behind representing and generating different RNA structural modalities, and propose a flexible framework to jointly embed and generate these molecular structures along with their sequence in a meaningful latent space. Equipped with a deep understanding of RNA molecular structures, our most sophisticated encoding and decoding methods operate on the molecular graph as well as the junction tree hierarchy, integrating strong inductive bias about RNA structural regularity and folding mechanism such that high structural validity, stability and diversity of generated RNAs are achieved. Also, we seek to adequately organize the latent space of RNA molecular embeddings with regard to the interaction with proteins, and targeted optimization is used to navigate in this latent space to search for desired novel RNA molecules.
Mycorrhiza: Genotype Assignment usingPhylogenetic Networks
Oct 14, 2020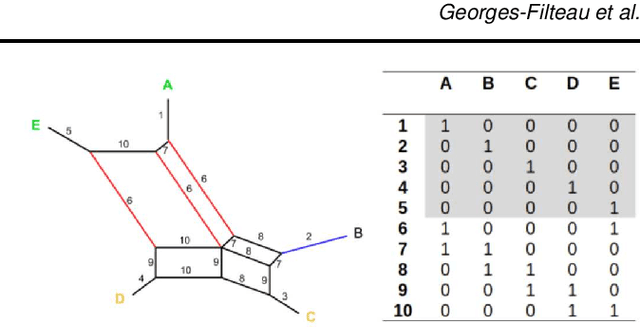
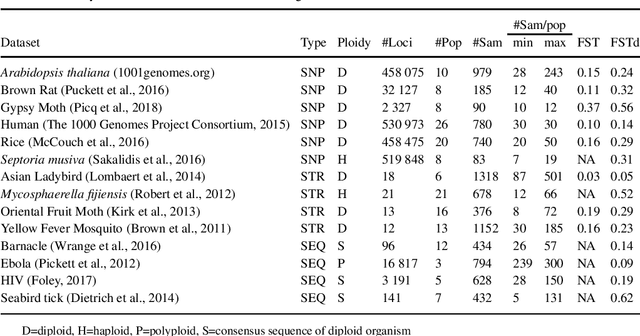
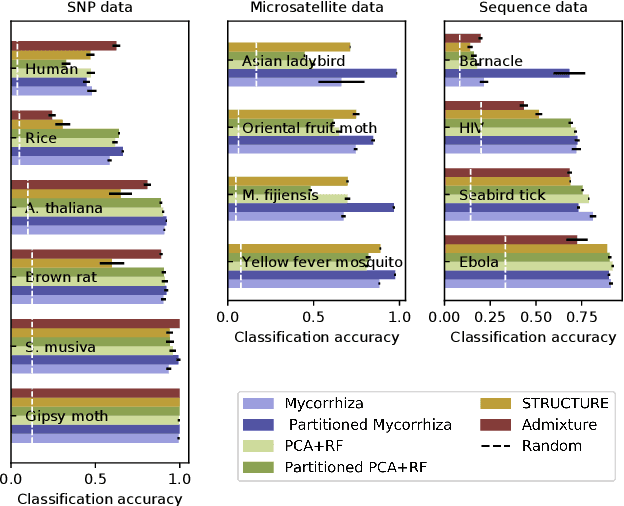
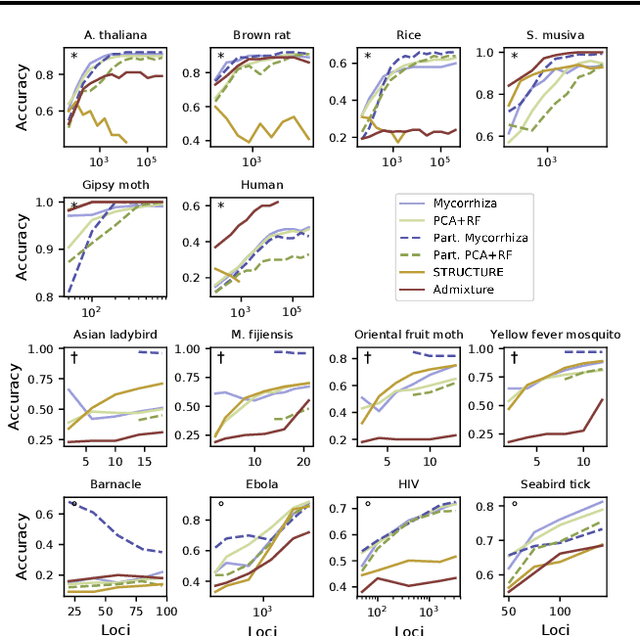
Motivation The genotype assignment problem consists of predicting, from the genotype of an individual, which of a known set of populations it originated from. The problem arises in a variety of contexts, including wildlife forensics, invasive species detection and biodiversity monitoring. Existing approaches perform well under ideal conditions but are sensitive to a variety of common violations of the assumptions they rely on. Results In this article, we introduce Mycorrhiza, a machine learning approach for the genotype assignment problem. Our algorithm makes use of phylogenetic networks to engineer features that encode the evolutionary relationships among samples. Those features are then used as input to a Random Forests classifier. The classification accuracy was assessed on multiple published empirical SNP, microsatellite or consensus sequence datasets with wide ranges of size, geographical distribution and population structure and on simulated datasets. It compared favorably against widely used assessment tests or mixture analysis methods such as STRUCTURE and Admixture, and against another machine-learning based approach using principal component analysis for dimensionality reduction. Mycorrhiza yields particularly significant gains on datasets with a large average fixation index (FST) or deviation from the Hardy-Weinberg equilibrium. Moreover, the phylogenetic network approach estimates mixture proportions with good accuracy.