 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning Where to See: A Novel Attention Model for Automated Immunohistochemical Scoring
Mar 26, 2019



Estimating over-amplification of human epidermal growth factor receptor 2 (HER2) on invasive breast cancer (BC) is regarded as a significant predictive and prognostic marker. We propose a novel deep reinforcement learning (DRL) based model that treats immunohistochemical (IHC) scoring of HER2 as a sequential learning task. For a given image tile sampled from multi-resolution giga-pixel whole slide image (WSI), the model learns to sequentially identify some of the diagnostically relevant regions of interest (ROIs) by following a parameterized policy. The selected ROIs are processed by recurrent and residual convolution networks to learn the discriminative features for different HER2 scores and predict the next location, without requiring to process all the sub-image patches of a given tile for predicting the HER2 score, mimicking the histopathologist who would not usually analyze every part of the slide at the highest magnification. The proposed model incorporates a task-specific regularization term and inhibition of return mechanism to prevent the model from revisiting the previously attended locations. We evaluated our model on two IHC datasets: a publicly available dataset from the HER2 scoring challenge contest and another dataset consisting of WSIs of gastroenteropancreatic neuroendocrine tumor sections stained with Glo1 marker. We demonstrate that the proposed model outperforms other methods based on state-of-the-art deep convolutional networks. To the best of our knowledge, this is the first study using DRL for IHC scoring and could potentially lead to wider use of DRL in the domain of computational pathology reducing the computational burden of the analysis of large multigigapixel histology images.
Methods for Segmentation and Classification of Digital Microscopy Tissue Images
Oct 31, 2018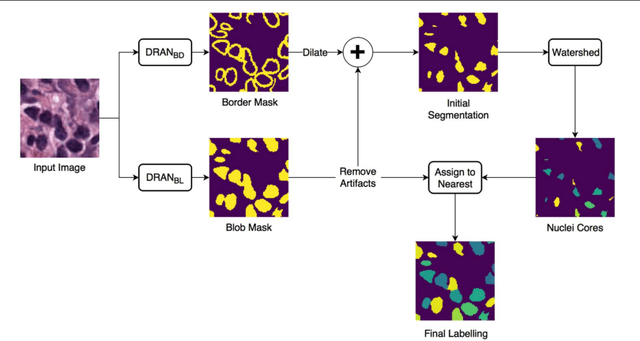

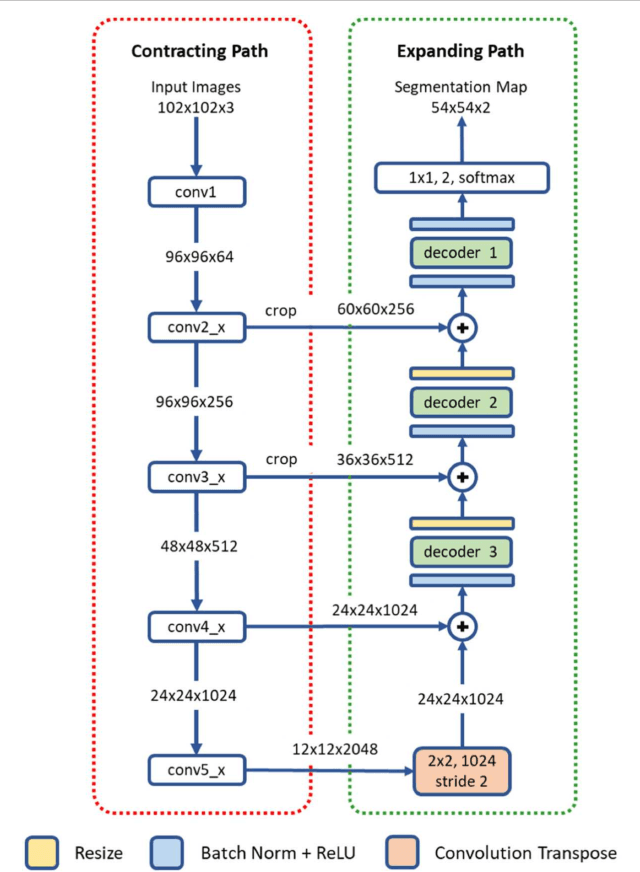
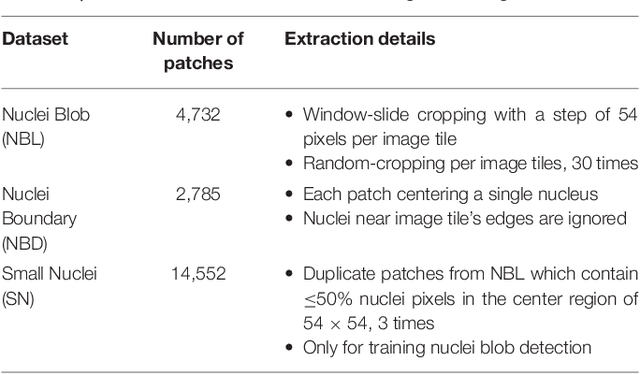
High-resolution microscopy images of tissue specimens provide detailed information about the morphology of normal and diseased tissue. Image analysis of tissue morphology can help cancer researchers develop a better understanding of cancer biology. Segmentation of nuclei and classification of tissue images are two common tasks in tissue image analysis. Development of accurate and efficient algorithms for these tasks is a challenging problem because of the complexity of tissue morphology and tumor heterogeneity. In this paper we present two computer algorithms; one designed for segmentation of nuclei and the other for classification of whole slide tissue images. The segmentation algorithm implements a multiscale deep residual aggregation network to accurately segment nuclear material and then separate clumped nuclei into individual nuclei. The classification algorithm initially carries out patch-level classification via a deep learning method, then patch-level statistical and morphological features are used as input to a random forest regression model for whole slide image classification. The segmentation and classification algorithms were evaluated in the MICCAI 2017 Digital Pathology challenge. The segmentation algorithm achieved an accuracy score of 0.78. The classification algorithm achieved an accuracy score of 0.81.
Leveraging Unlabeled Whole-Slide-Images for Mitosis Detection
Jul 31, 2018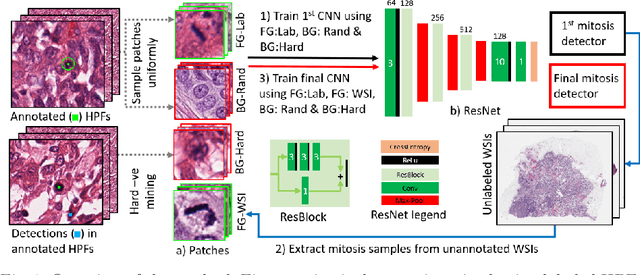
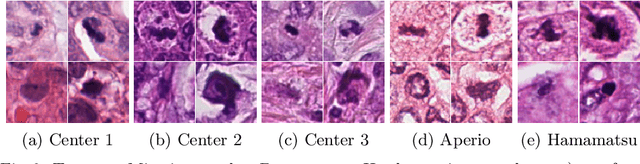
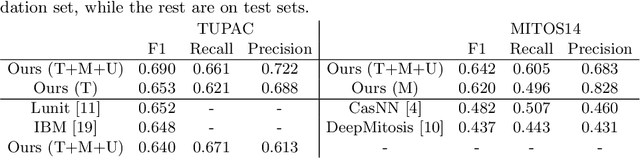
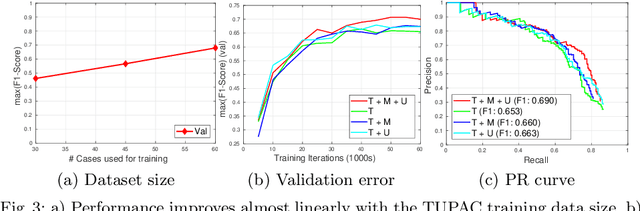
Mitosis count is an important biomarker for prognosis of various cancers. At present, pathologists typically perform manual counting on a few selected regions of interest in breast whole-slide-images (WSIs) of patient biopsies. This task is very time-consuming, tedious and subjective. Automated mitosis detection methods have made great advances in recent years. However, these methods require exhaustive labeling of a large number of selected regions of interest. This task is very expensive because expert pathologists are needed for reliable and accurate annotations. In this paper, we present a semi-supervised mitosis detection method which is designed to leverage a large number of unlabeled breast cancer WSIs. As a result, our method capitalizes on the growing number of digitized histology images, without relying on exhaustive annotations, subsequently improving mitosis detection. Our method first learns a mitosis detector from labeled data, uses this detector to mine additional mitosis samples from unlabeled WSIs, and then trains the final model using this larger and diverse set of mitosis samples. The use of unlabeled data improves F1-score by $\sim$5\% compared to our best performing fully-supervised model on the TUPAC validation set. Our submission (single model) to TUPAC challenge ranks highly on the leaderboard with an F1-score of 0.64.
Predicting breast tumor proliferation from whole-slide images: the TUPAC16 challenge
Jul 22, 2018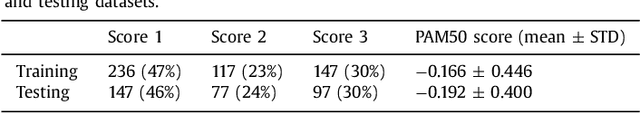
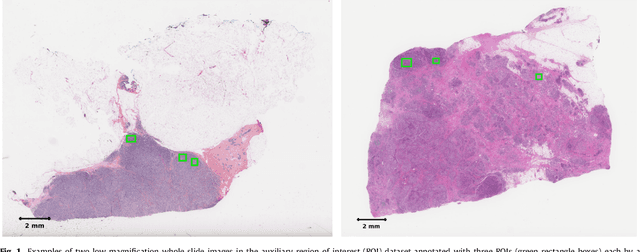
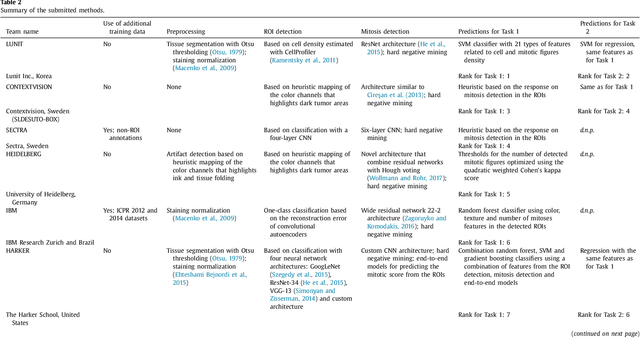
Tumor proliferation is an important biomarker indicative of the prognosis of breast cancer patients. Assessment of tumor proliferation in a clinical setting is highly subjective and labor-intensive task. Previous efforts to automate tumor proliferation assessment by image analysis only focused on mitosis detection in predefined tumor regions. However, in a real-world scenario, automatic mitosis detection should be performed in whole-slide images (WSIs) and an automatic method should be able to produce a tumor proliferation score given a WSI as input. To address this, we organized the TUmor Proliferation Assessment Challenge 2016 (TUPAC16) on prediction of tumor proliferation scores from WSIs. The challenge dataset consisted of 500 training and 321 testing breast cancer histopathology WSIs. In order to ensure fair and independent evaluation, only the ground truth for the training dataset was provided to the challenge participants. The first task of the challenge was to predict mitotic scores, i.e., to reproduce the manual method of assessing tumor proliferation by a pathologist. The second task was to predict the gene expression based PAM50 proliferation scores from the WSI. The best performing automatic method for the first task achieved a quadratic-weighted Cohen's kappa score of $\kappa$ = 0.567, 95% CI [0.464, 0.671] between the predicted scores and the ground truth. For the second task, the predictions of the top method had a Spearman's correlation coefficient of r = 0.617, 95% CI [0.581 0.651] with the ground truth. This was the first study that investigated tumor proliferation assessment from WSIs. The achieved results are promising given the difficulty of the tasks and weakly-labelled nature of the ground truth. However, further research is needed to improve the practical utility of image analysis methods for this task.
Fast and Accurate Tumor Segmentation of Histology Images using Persistent Homology and Deep Convolutional Features
May 09, 2018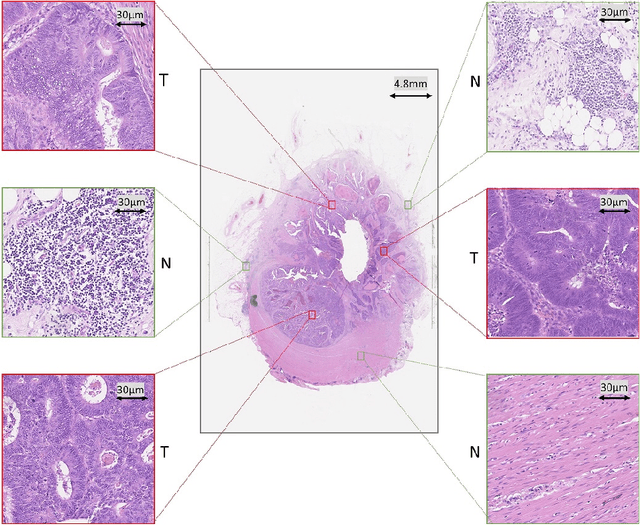
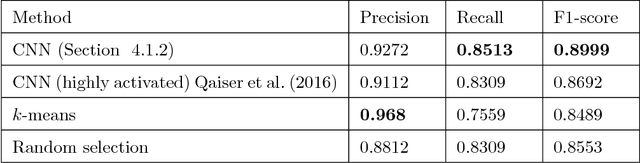

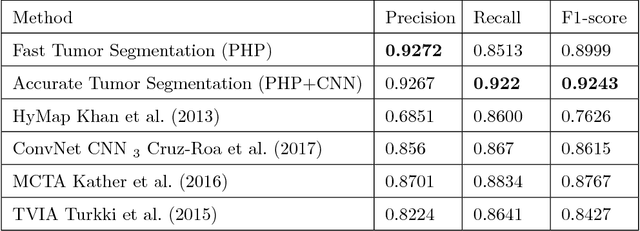
Tumor segmentation in whole-slide images of histology slides is an important step towards computer-assisted diagnosis. In this work, we propose a tumor segmentation framework based on the novel concept of persistent homology profiles (PHPs). For a given image patch, the homology profiles are derived by efficient computation of persistent homology, which is an algebraic tool from homology theory. We propose an efficient way of computing topological persistence of an image, alternative to simplicial homology. The PHPs are devised to distinguish tumor regions from their normal counterparts by modeling the atypical characteristics of tumor nuclei. We propose two variants of our method for tumor segmentation: one that targets speed without compromising accuracy and the other that targets higher accuracy. The fast version is based on the selection of exemplar image patches from a convolution neural network (CNN) and patch classification by quantifying the divergence between the PHPs of exemplars and the input image patch. Detailed comparative evaluation shows that the proposed algorithm is significantly faster than competing algorithms while achieving comparable results. The accurate version combines the PHPs and high-level CNN features and employs a multi-stage ensemble strategy for image patch labeling. Experimental results demonstrate that the combination of PHPs and CNN features outperforms competing algorithms. This study is performed on two independently collected colorectal datasets containing adenoma, adenocarcinoma, signet and healthy cases. Collectively, the accurate tumor segmentation produces the highest average patch-level F1-score, as compared with competing algorithms, on malignant and healthy cases from both the datasets. Overall the proposed framework highlights the utility of persistent homology for histopathology image analysis.
Her2 Challenge Contest: A Detailed Assessment of Automated Her2 Scoring Algorithms in Whole Slide Images of Breast Cancer Tissues
Jul 24, 2017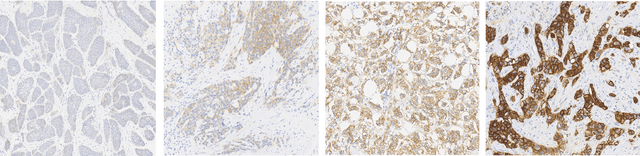

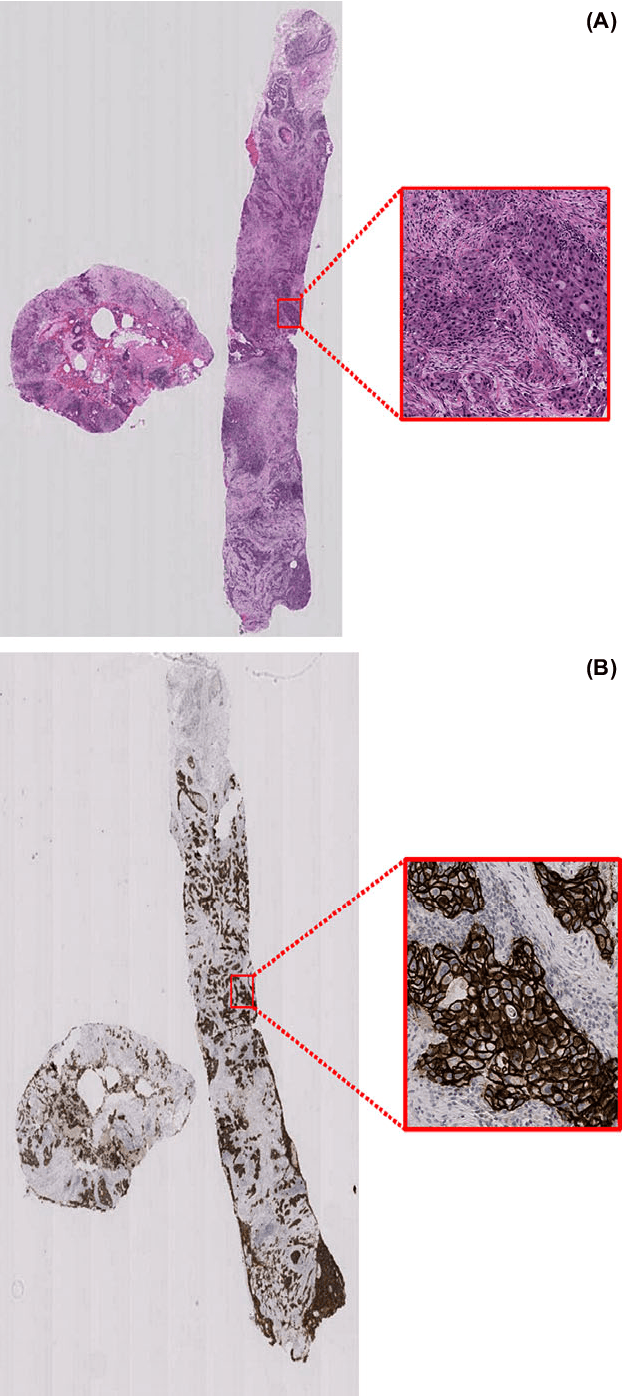
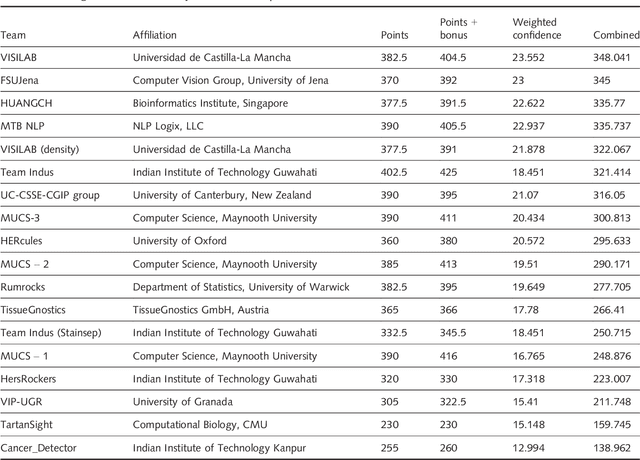
Evaluating expression of the Human epidermal growth factor receptor 2 (Her2) by visual examination of immunohistochemistry (IHC) on invasive breast cancer (BCa) is a key part of the diagnostic assessment of BCa due to its recognised importance as a predictive and prognostic marker in clinical practice. However, visual scoring of Her2 is subjective and consequently prone to inter-observer variability. Given the prognostic and therapeutic implications of Her2 scoring, a more objective method is required. In this paper, we report on a recent automated Her2 scoring contest, held in conjunction with the annual PathSoc meeting held in Nottingham in June 2016, aimed at systematically comparing and advancing the state-of-the-art Artificial Intelligence (AI) based automated methods for Her2 scoring. The contest dataset comprised of digitised whole slide images (WSI) of sections from 86 cases of invasive breast carcinoma stained with both Haematoxylin & Eosin (H&E) and IHC for Her2. The contesting algorithms automatically predicted scores of the IHC slides for an unseen subset of the dataset and the predicted scores were compared with the 'ground truth' (a consensus score from at least two experts). We also report on a simple Man vs Machine contest for the scoring of Her2 and show that the automated methods could beat the pathology experts on this contest dataset. This paper presents a benchmark for comparing the performance of automated algorithms for scoring of Her2. It also demonstrates the enormous potential of automated algorithms in assisting the pathologist with objective IHC scoring.