 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCommunicative Subgraph Representation Learning for Multi-Relational Inductive Drug-Gene Interaction Prediction
May 12, 2022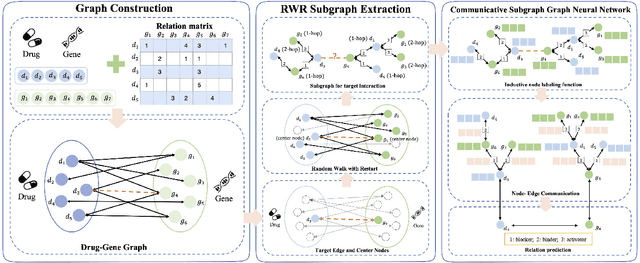
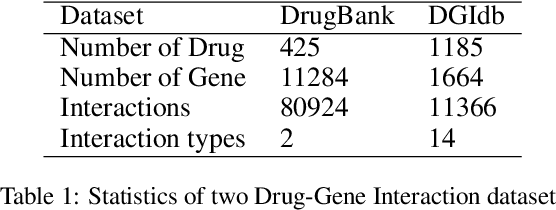
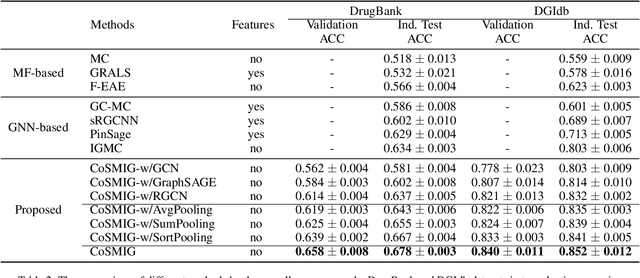
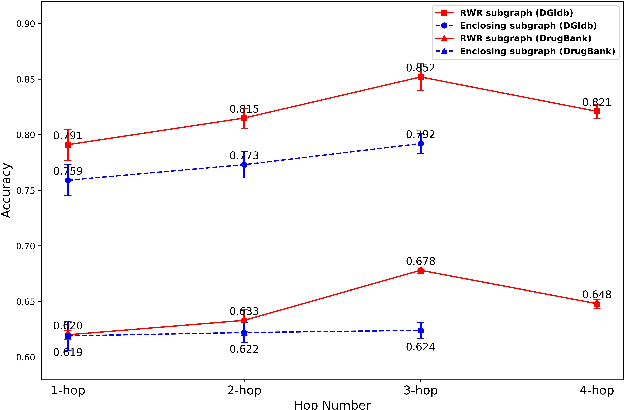
Illuminating the interconnections between drugs and genes is an important topic in drug development and precision medicine. Currently, computational predictions of drug-gene interactions mainly focus on the binding interactions without considering other relation types like agonist, antagonist, etc. In addition, existing methods either heavily rely on high-quality domain features or are intrinsically transductive, which limits the capacity of models to generalize to drugs/genes that lack external information or are unseen during the training process. To address these problems, we propose a novel Communicative Subgraph representation learning for Multi-relational Inductive drug-Gene interactions prediction (CoSMIG), where the predictions of drug-gene relations are made through subgraph patterns, and thus are naturally inductive for unseen drugs/genes without retraining or utilizing external domain features. Moreover, the model strengthened the relations on the drug-gene graph through a communicative message passing mechanism. To evaluate our method, we compiled two new benchmark datasets from DrugBank and DGIdb. The comprehensive experiments on the two datasets showed that our method outperformed state-of-the-art baselines in the transductive scenarios and achieved superior performance in the inductive ones. Further experimental analysis including LINCS experimental validation and literature verification also demonstrated the value of our model.
Molecular Attributes Transfer from Non-Parallel Data
Nov 30, 2021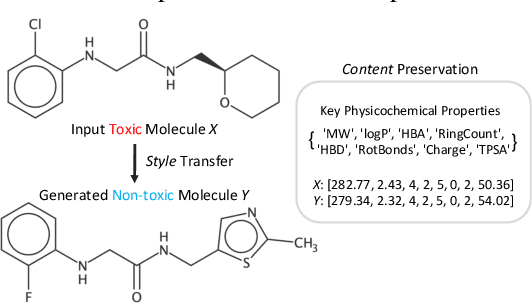
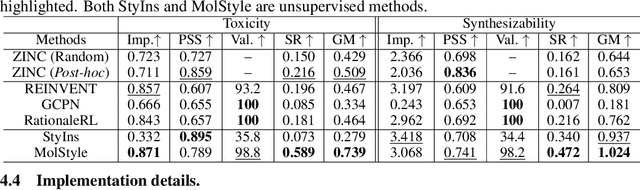
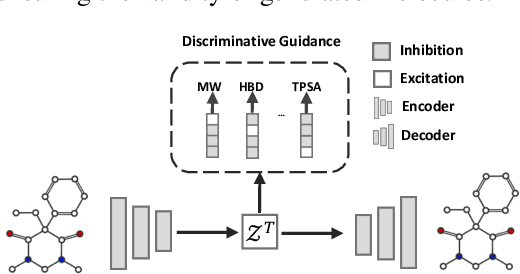
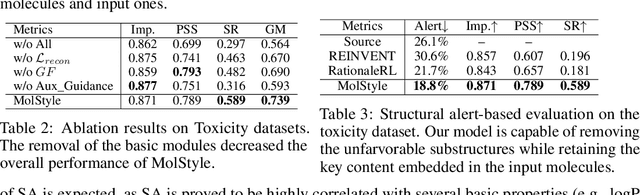
Optimizing chemical molecules for desired properties lies at the core of drug development. Despite initial successes made by deep generative models and reinforcement learning methods, these methods were mostly limited by the requirement of predefined attribute functions or parallel data with manually pre-compiled pairs of original and optimized molecules. In this paper, for the first time, we formulate molecular optimization as a style transfer problem and present a novel generative model that could automatically learn internal differences between two groups of non-parallel data through adversarial training strategies. Our model further enables both preservation of molecular contents and optimization of molecular properties through combining auxiliary guided-variational autoencoders and generative flow techniques. Experiments on two molecular optimization tasks, toxicity modification and synthesizability improvement, demonstrate that our model significantly outperforms several state-of-the-art methods.
Hybrid Contrastive Learning of Tri-Modal Representation for Multimodal Sentiment Analysis
Sep 04, 2021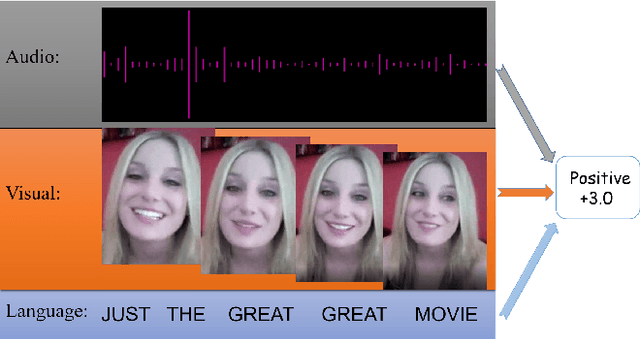
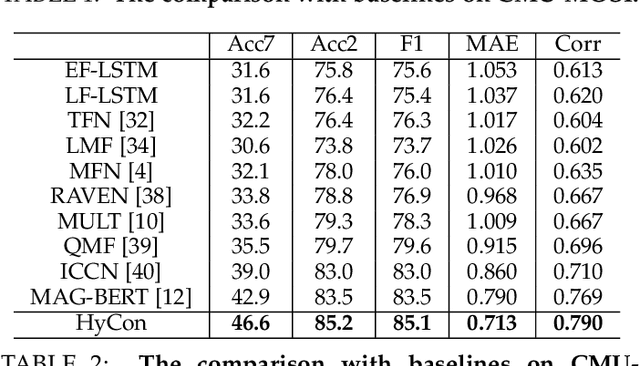
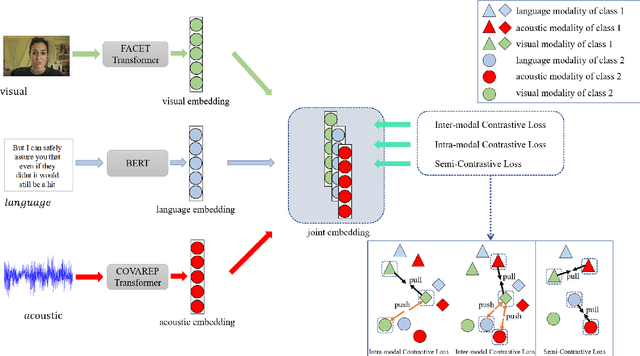
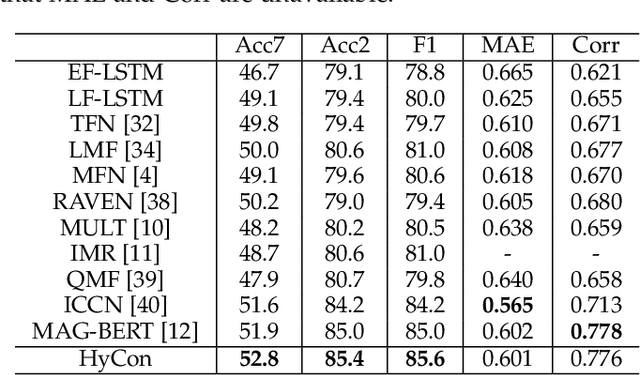
The wide application of smart devices enables the availability of multimodal data, which can be utilized in many tasks. In the field of multimodal sentiment analysis (MSA), most previous works focus on exploring intra- and inter-modal interactions. However, training a network with cross-modal information (language, visual, audio) is still challenging due to the modality gap, and existing methods still cannot ensure to sufficiently learn intra-/inter-modal dynamics. Besides, while learning dynamics within each sample draws great attention, the learning of inter-class relationships is neglected. Moreover, the size of datasets limits the generalization ability of existing methods. To address the afore-mentioned issues, we propose a novel framework HyCon for hybrid contrastive learning of tri-modal representation. Specifically, we simultaneously perform intra-/inter-modal contrastive learning and semi-contrastive learning (that is why we call it hybrid contrastive learning), with which the model can fully explore cross-modal interactions, preserve inter-class relationships and reduce the modality gap. Besides, a refinement term is devised to prevent the model falling into a sub-optimal solution. Moreover, HyCon can naturally generate a large amount of training pairs for better generalization and reduce the negative effect of limited datasets. Extensive experiments on public datasets demonstrate that our proposed method outperforms existing works.
Learning Attributed Graph Representations with Communicative Message Passing Transformer
Jul 28, 2021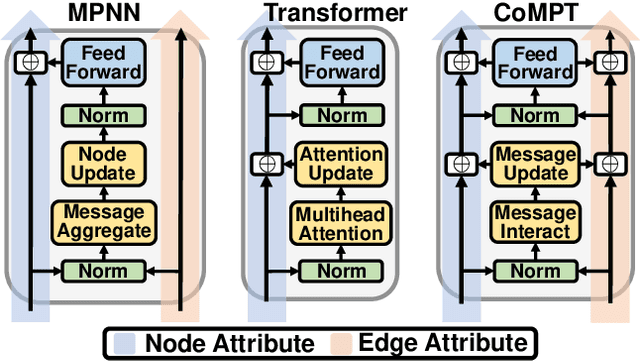
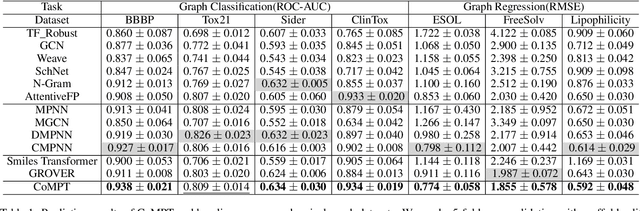
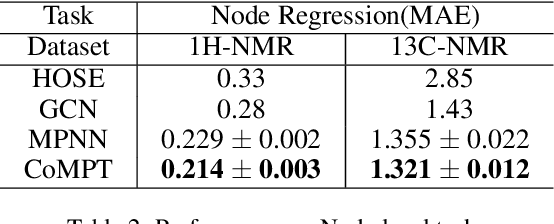
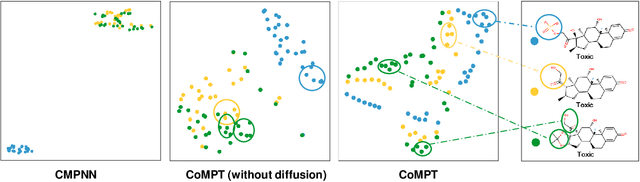
Constructing appropriate representations of molecules lies at the core of numerous tasks such as material science, chemistry and drug designs. Recent researches abstract molecules as attributed graphs and employ graph neural networks (GNN) for molecular representation learning, which have made remarkable achievements in molecular graph modeling. Albeit powerful, current models either are based on local aggregation operations and thus miss higher-order graph properties or focus on only node information without fully using the edge information. For this sake, we propose a Communicative Message Passing Transformer (CoMPT) neural network to improve the molecular graph representation by reinforcing message interactions between nodes and edges based on the Transformer architecture. Unlike the previous transformer-style GNNs that treat molecules as fully connected graphs, we introduce a message diffusion mechanism to leverage the graph connectivity inductive bias and reduce the message enrichment explosion. Extensive experiments demonstrated that the proposed model obtained superior performances (around 4$\%$ on average) against state-of-the-art baselines on seven chemical property datasets (graph-level tasks) and two chemical shift datasets (node-level tasks). Further visualization studies also indicated a better representation capacity achieved by our model.
Subgraph-aware Few-Shot Inductive Link Prediction via Meta-Learning
Jul 26, 2021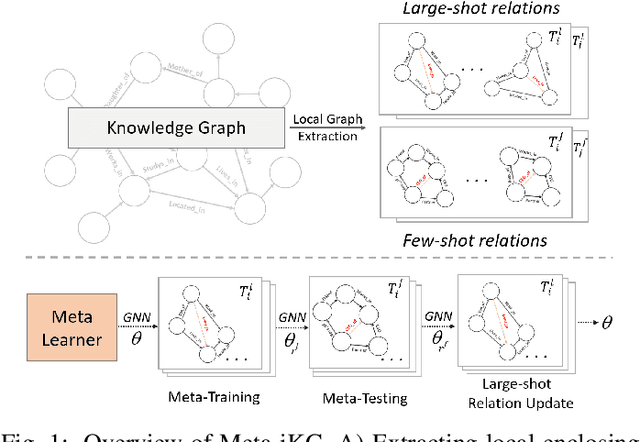
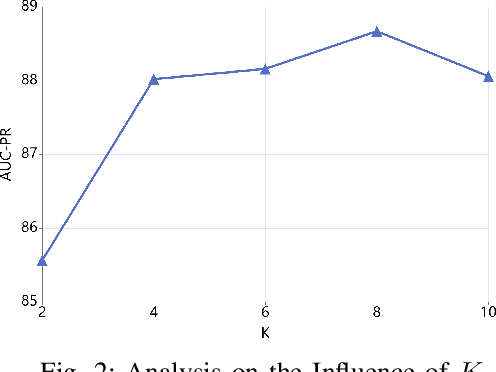
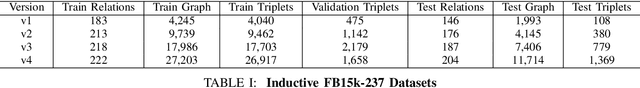
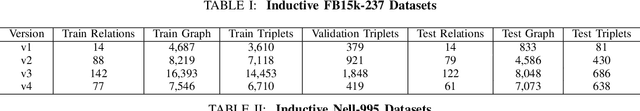
Link prediction for knowledge graphs aims to predict missing connections between entities. Prevailing methods are limited to a transductive setting and hard to process unseen entities. The recent proposed subgraph-based models provided alternatives to predict links from the subgraph structure surrounding a candidate triplet. However, these methods require abundant known facts of training triplets and perform poorly on relationships that only have a few triplets. In this paper, we propose Meta-iKG, a novel subgraph-based meta-learner for few-shot inductive relation reasoning. Meta-iKG utilizes local subgraphs to transfer subgraph-specific information and learn transferable patterns faster via meta gradients. In this way, we find the model can quickly adapt to few-shot relationships using only a handful of known facts with inductive settings. Moreover, we introduce a large-shot relation update procedure to traditional meta-learning to ensure that our model can generalize well both on few-shot and large-shot relations. We evaluate Meta-iKG on inductive benchmarks sampled from NELL and Freebase, and the results show that Meta-iKG outperforms the current state-of-the-art methods both in few-shot scenarios and standard inductive settings.
Quantitative Evaluation of Explainable Graph Neural Networks for Molecular Property Prediction
Jul 12, 2021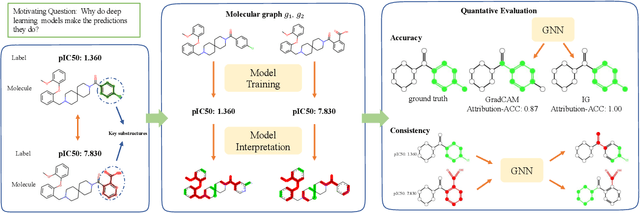
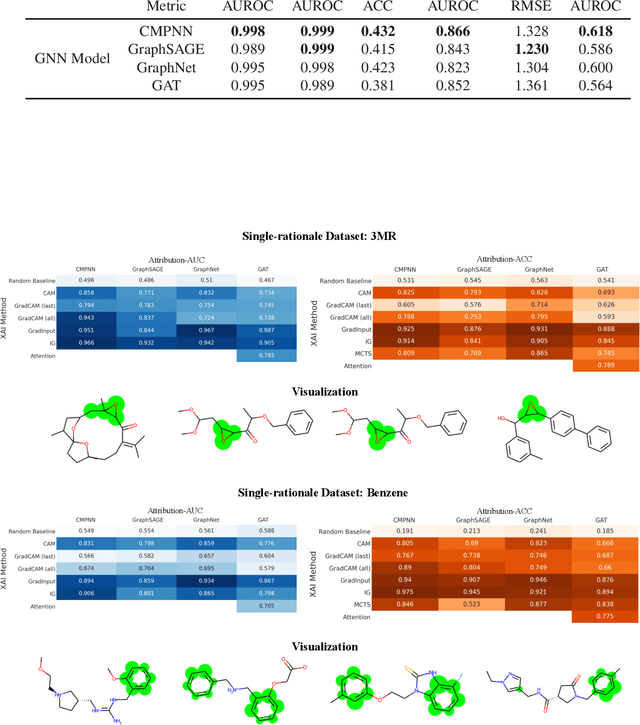
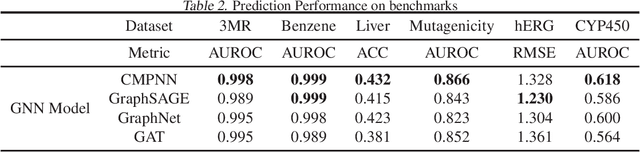
Advances in machine learning have led to graph neural network-based methods for drug discovery, yielding promising results in molecular design, chemical synthesis planning, and molecular property prediction. However, current graph neural networks (GNNs) remain of limited acceptance in drug discovery is limited due to their lack of interpretability. Although this major weakness has been mitigated by the development of explainable artificial intelligence (XAI) techniques, the "ground truth" assignment in most explainable tasks ultimately rests with subjective judgments by humans so that the quality of model interpretation is hard to evaluate in quantity. In this work, we first build three levels of benchmark datasets to quantitatively assess the interpretability of the state-of-the-art GNN models. Then we implemented recent XAI methods in combination with different GNN algorithms to highlight the benefits, limitations, and future opportunities for drug discovery. As a result, GradInput and IG generally provide the best model interpretability for GNNs, especially when combined with GraphNet and CMPNN. The integrated and developed XAI package is fully open-sourced and can be used by practitioners to train new models on other drug discovery tasks.
BioNavi-NP: Biosynthesis Navigator for Natural Products
May 26, 2021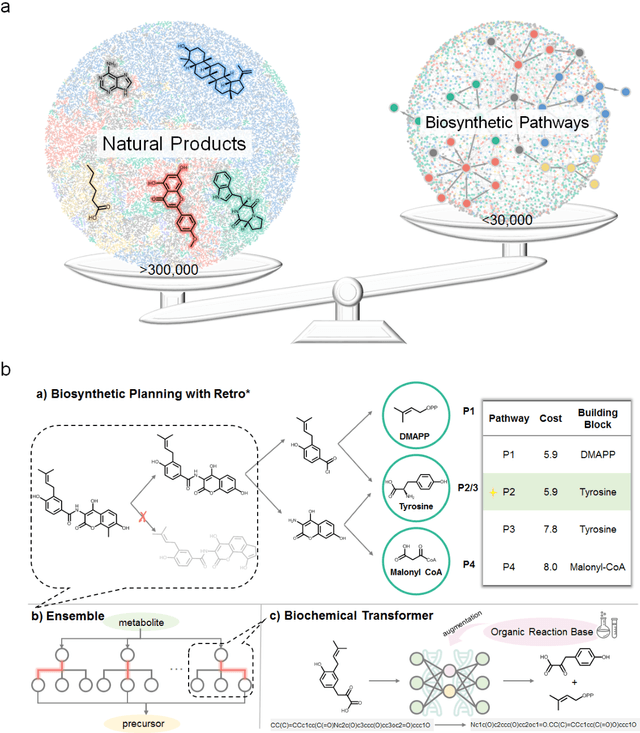
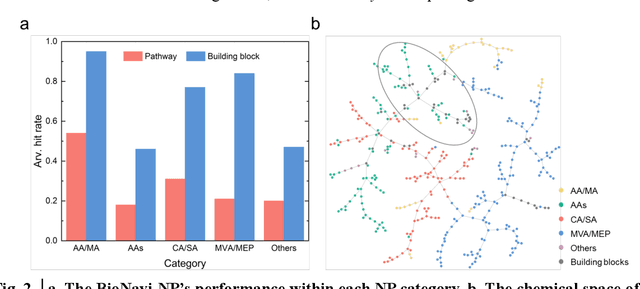
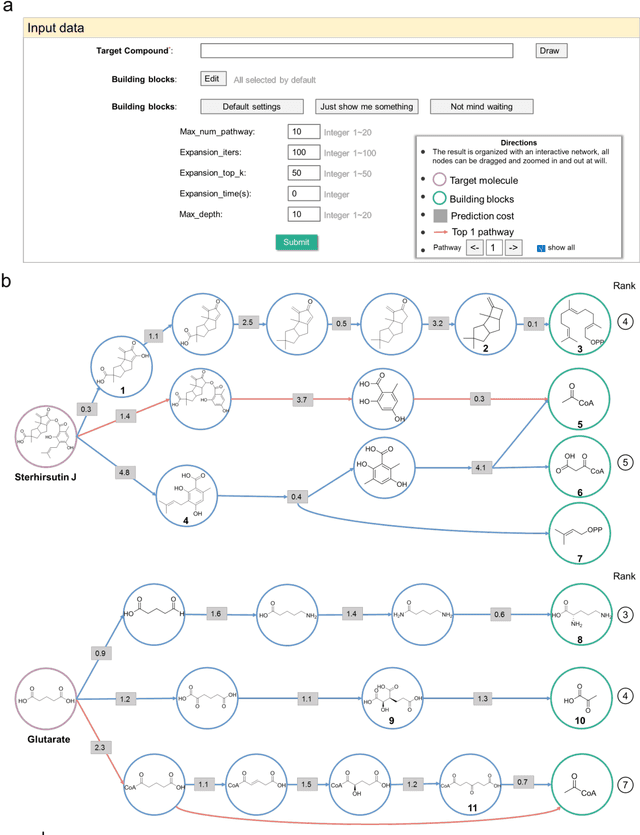
Nature, a synthetic master, creates more than 300,000 natural products (NPs) which are the major constituents of FDA-proved drugs owing to the vast chemical space of NPs. To date, there are fewer than 30,000 validated NPs compounds involved in about 33,000 known enzyme catalytic reactions, and even fewer biosynthetic pathways are known with complete cascade-connected enzyme catalysis. Therefore, it is valuable to make computer-aided bio-retrosynthesis predictions. Here, we develop BioNavi-NP, a navigable and user-friendly toolkit, which is capable of predicting the biosynthetic pathways for NPs and NP-like compounds through a novel (AND-OR Tree)-based planning algorithm, an enhanced molecular Transformer neural network, and a training set that combines general organic transformations and biosynthetic steps. Extensive evaluations reveal that BioNavi-NP generalizes well to identifying the reported biosynthetic pathways for 90% of test compounds and recovering the verified building blocks for 73%, significantly outperforming conventional rule-based approaches. Moreover, BioNavi-NP also shows an outstanding capacity of biologically plausible pathways enumeration. In this sense, BioNavi-NP is a leading-edge toolkit to redesign complex biosynthetic pathways of natural products with applications to total or semi-synthesis and pathway elucidation or reconstruction.
Communicative Message Passing for Inductive Relation Reasoning
Dec 16, 2020



Relation prediction for knowledge graphs aims at predicting missing relationships between entities. Despite the importance of inductive relation prediction, most previous works are limited to a transductive setting and cannot process previously unseen entities. The recent proposed subgraph-based relation reasoning models provided alternatives to predict links from the subgraph structure surrounding a candidate triplet inductively. However, we observe that these methods often neglect the directed nature of the extracted subgraph and weaken the role of relation information in the subgraph modeling. As a result, they fail to effectively handle the asymmetric/anti-symmetric triplets and produce insufficient embeddings for the target triplets. To this end, we introduce a \textbf{C}\textbf{o}mmunicative \textbf{M}essage \textbf{P}assing neural network for \textbf{I}nductive re\textbf{L}ation r\textbf{E}asoning, \textbf{CoMPILE}, that reasons over local directed subgraph structures and has a vigorous inductive bias to process entity-independent semantic relations. In contrast to existing models, CoMPILE strengthens the message interactions between edges and entitles through a communicative kernel and enables a sufficient flow of relation information. Moreover, we demonstrate that CoMPILE can naturally handle asymmetric/anti-symmetric relations without the need for explosively increasing the number of model parameters by extracting the directed enclosing subgraphs. Extensive experiments show substantial performance gains in comparison to state-of-the-art methods on commonly used benchmark datasets with variant inductive settings.
RetroXpert: Decompose Retrosynthesis Prediction like a Chemist
Nov 04, 2020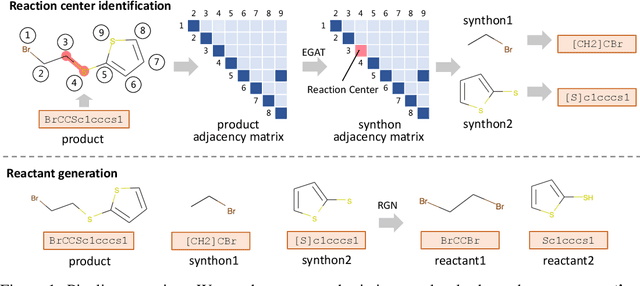
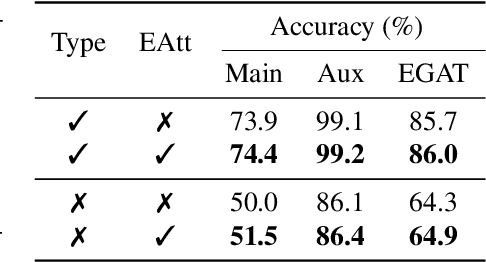
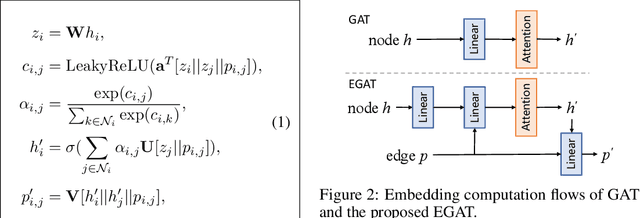
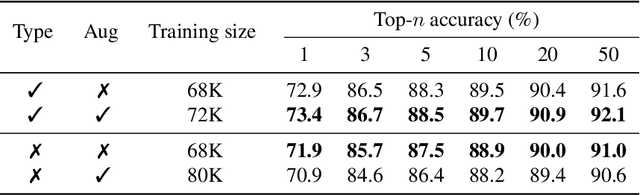
Retrosynthesis is the process of recursively decomposing target molecules into available building blocks. It plays an important role in solving problems in organic synthesis planning. To automate or assist in the retrosynthesis analysis, various retrosynthesis prediction algorithms have been proposed. However, most of them are cumbersome and lack interpretability about their predictions. In this paper, we devise a novel template-free algorithm for automatic retrosynthetic expansion inspired by how chemists approach retrosynthesis prediction. Our method disassembles retrosynthesis into two steps: i) identify the potential reaction center of the target molecule through a novel graph neural network and generate intermediate synthons, and ii) generate the reactants associated with synthons via a robust reactant generation model. While outperforming the state-of-the-art baselines by a significant margin, our model also provides chemically reasonable interpretation.
Predicting Retrosynthetic Reaction using Self-Corrected Transformer Neural Networks
Jul 03, 2019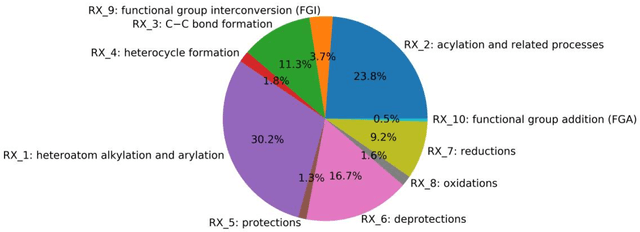
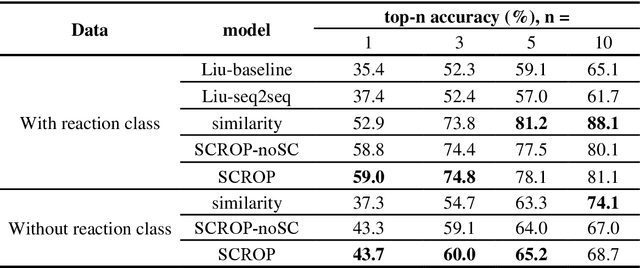
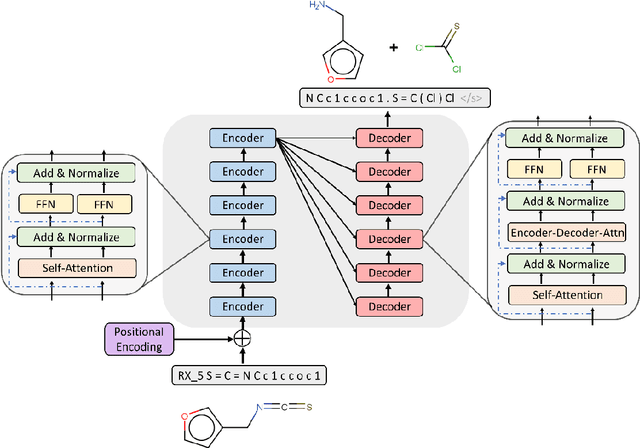
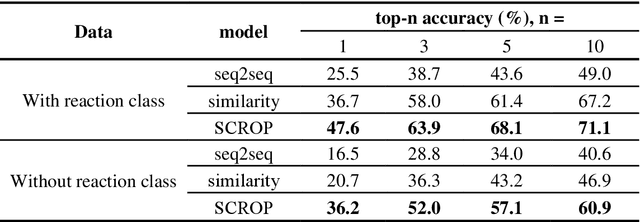
Synthesis planning is the process of recursively decomposing target molecules into available precursors. Computer-aided retrosynthesis can potentially assist chemists in designing synthetic routes, but at present it is cumbersome and provides results of dissatisfactory quality. In this study, we develop a template-free self-corrected retrosynthesis predictor (SCROP) to perform a retrosynthesis prediction task trained by using the Transformer neural network architecture. In the method, the retrosynthesis planning is converted as a machine translation problem between molecular linear notations of reactants and the products. Coupled with a neural network-based syntax corrector, our method achieves an accuracy of 59.0% on a standard benchmark dataset, which increases >21% over other deep learning methods, and >6% over template-based methods. More importantly, our method shows an accuracy 1.7 times higher than other state-of-the-art methods for compounds not appearing in the training set.