 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBiomedical image analysis competitions: The state of current participation practice
Dec 16, 2022The number of international benchmarking competitions is steadily increasing in various fields of machine learning (ML) research and practice. So far, however, little is known about the common practice as well as bottlenecks faced by the community in tackling the research questions posed. To shed light on the status quo of algorithm development in the specific field of biomedical imaging analysis, we designed an international survey that was issued to all participants of challenges conducted in conjunction with the IEEE ISBI 2021 and MICCAI 2021 conferences (80 competitions in total). The survey covered participants' expertise and working environments, their chosen strategies, as well as algorithm characteristics. A median of 72% challenge participants took part in the survey. According to our results, knowledge exchange was the primary incentive (70%) for participation, while the reception of prize money played only a minor role (16%). While a median of 80 working hours was spent on method development, a large portion of participants stated that they did not have enough time for method development (32%). 25% perceived the infrastructure to be a bottleneck. Overall, 94% of all solutions were deep learning-based. Of these, 84% were based on standard architectures. 43% of the respondents reported that the data samples (e.g., images) were too large to be processed at once. This was most commonly addressed by patch-based training (69%), downsampling (37%), and solving 3D analysis tasks as a series of 2D tasks. K-fold cross-validation on the training set was performed by only 37% of the participants and only 50% of the participants performed ensembling based on multiple identical models (61%) or heterogeneous models (39%). 48% of the respondents applied postprocessing steps.
Mitosis domain generalization in histopathology images -- The MIDOG challenge
Apr 06, 2022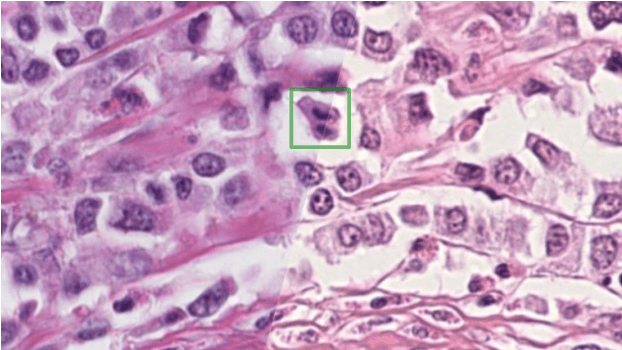
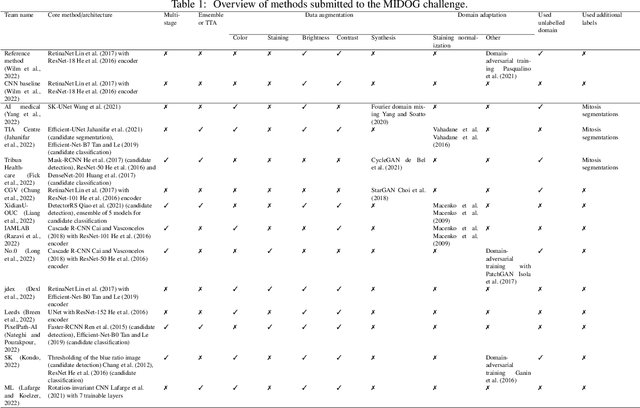
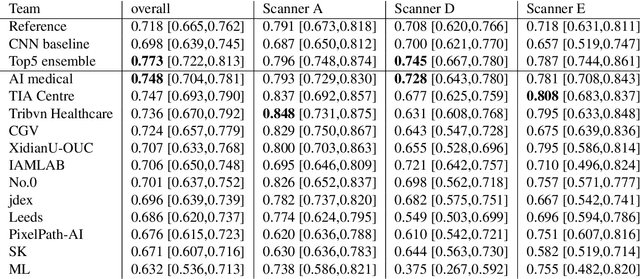
The density of mitotic figures within tumor tissue is known to be highly correlated with tumor proliferation and thus is an important marker in tumor grading. Recognition of mitotic figures by pathologists is known to be subject to a strong inter-rater bias, which limits the prognostic value. State-of-the-art deep learning methods can support the expert in this assessment but are known to strongly deteriorate when applied in a different clinical environment than was used for training. One decisive component in the underlying domain shift has been identified as the variability caused by using different whole slide scanners. The goal of the MICCAI MIDOG 2021 challenge has been to propose and evaluate methods that counter this domain shift and derive scanner-agnostic mitosis detection algorithms. The challenge used a training set of 200 cases, split across four scanning systems. As a test set, an additional 100 cases split across four scanning systems, including two previously unseen scanners, were given. The best approaches performed on an expert level, with the winning algorithm yielding an F_1 score of 0.748 (CI95: 0.704-0.781). In this paper, we evaluate and compare the approaches that were submitted to the challenge and identify methodological factors contributing to better performance.
Cascade RCNN for MIDOG Challenge
Sep 26, 2021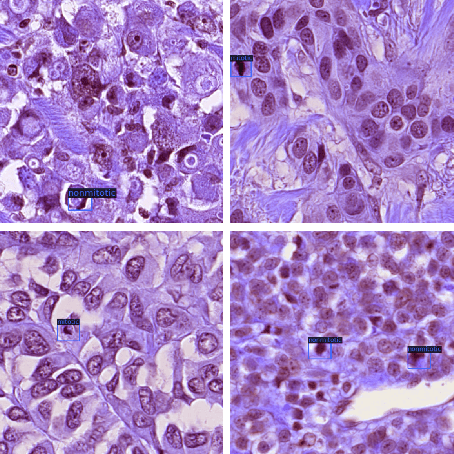
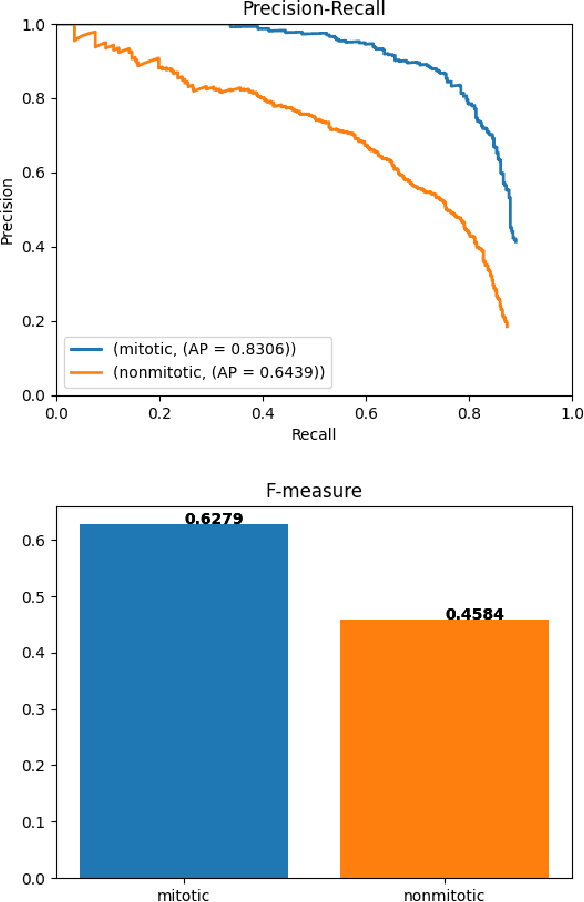
Mitotic counts are one of the key indicators of breast cancer prognosis. However, accurate mitotic cell counting is still a difficult problem and is labourious. Automated methods have been proposed for this task, but are usually dependent on the training images and show poor performance on unseen domains. In this work, we present a multi-stage mitosis detection method based on a Cascade RCNN developed to be sequentially more selective against false positives. On the preliminary test set, the algorithm scores an F1-score of 0.7492.
Mitosis Detection for Breast Cancer Pathology Images using UV-Net
Sep 21, 2021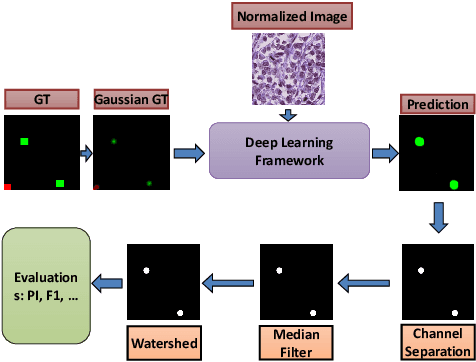
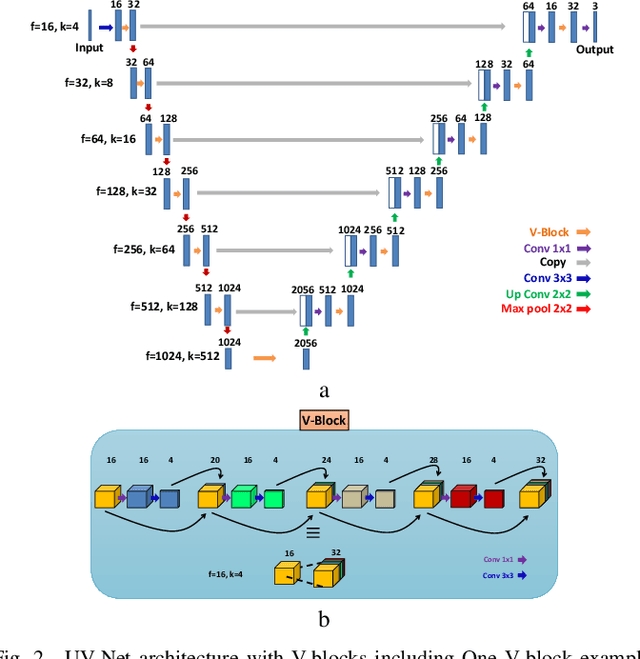
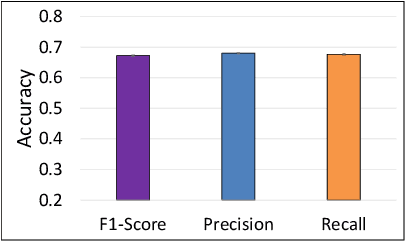
The difficulty of detecting mitosis and its similarity to non-mitosis objects has remained a challenge in computational pathology. The lack of publicly available data has added more complexity. Deep learning algorithms have shown potentials in mitosis detection tasks. However, they face challenges when applied to pathology images with dense medium and diverse dataset. This paper introduces an optimized UV-Net architecture, developed to focus on mitosis details with high-resolution through feature preservation. Stain normalization methods are used to generalize the trained network. An F1 score of 0.6721 is achieved using this network.