 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn Effective Meaningful Way to Evaluate Survival Models
Jun 01, 2023One straightforward metric to evaluate a survival prediction model is based on the Mean Absolute Error (MAE) -- the average of the absolute difference between the time predicted by the model and the true event time, over all subjects. Unfortunately, this is challenging because, in practice, the test set includes (right) censored individuals, meaning we do not know when a censored individual actually experienced the event. In this paper, we explore various metrics to estimate MAE for survival datasets that include (many) censored individuals. Moreover, we introduce a novel and effective approach for generating realistic semi-synthetic survival datasets to facilitate the evaluation of metrics. Our findings, based on the analysis of the semi-synthetic datasets, reveal that our proposed metric (MAE using pseudo-observations) is able to rank models accurately based on their performance, and often closely matches the true MAE -- in particular, is better than several alternative methods.
The ACROBAT 2022 Challenge: Automatic Registration Of Breast Cancer Tissue
May 29, 2023The alignment of tissue between histopathological whole-slide-images (WSI) is crucial for research and clinical applications. Advances in computing, deep learning, and availability of large WSI datasets have revolutionised WSI analysis. Therefore, the current state-of-the-art in WSI registration is unclear. To address this, we conducted the ACROBAT challenge, based on the largest WSI registration dataset to date, including 4,212 WSIs from 1,152 breast cancer patients. The challenge objective was to align WSIs of tissue that was stained with routine diagnostic immunohistochemistry to its H&E-stained counterpart. We compare the performance of eight WSI registration algorithms, including an investigation of the impact of different WSI properties and clinical covariates. We find that conceptually distinct WSI registration methods can lead to highly accurate registration performances and identify covariates that impact performances across methods. These results establish the current state-of-the-art in WSI registration and guide researchers in selecting and developing methods.
Modeling and Forecasting COVID-19 Cases using Latent Subpopulations
Feb 09, 2023Classical epidemiological models assume homogeneous populations. There have been important extensions to model heterogeneous populations, when the identity of the sub-populations is known, such as age group or geographical location. Here, we propose two new methods to model the number of people infected with COVID-19 over time, each as a linear combination of latent sub-populations -- i.e., when we do not know which person is in which sub-population, and the only available observations are the aggregates across all sub-populations. Method #1 is a dictionary-based approach, which begins with a large number of pre-defined sub-population models (each with its own starting time, shape, etc), then determines the (positive) weight of small (learned) number of sub-populations. Method #2 is a mixture-of-$M$ fittable curves, where $M$, the number of sub-populations to use, is given by the user. Both methods are compatible with any parametric model; here we demonstrate their use with first (a)~Gaussian curves and then (b)~SIR trajectories. We empirically show the performance of the proposed methods, first in (i) modeling the observed data and then in (ii) forecasting the number of infected people 1 to 4 weeks in advance. Across 187 countries, we show that the dictionary approach had the lowest mean absolute percentage error and also the lowest variance when compared with classical SIR models and moreover, it was a strong baseline that outperforms many of the models developed for COVID-19 forecasting.
Improving ECG-based COVID-19 diagnosis and mortality predictions using pre-pandemic medical records at population-scale
Nov 14, 2022



Pandemic outbreaks such as COVID-19 occur unexpectedly, and need immediate action due to their potential devastating consequences on global health. Point-of-care routine assessments such as electrocardiogram (ECG), can be used to develop prediction models for identifying individuals at risk. However, there is often too little clinically-annotated medical data, especially in early phases of a pandemic, to develop accurate prediction models. In such situations, historical pre-pandemic health records can be utilized to estimate a preliminary model, which can then be fine-tuned based on limited available pandemic data. This study shows this approach -- pre-train deep learning models with pre-pandemic data -- can work effectively, by demonstrating substantial performance improvement over three different COVID-19 related diagnostic and prognostic prediction tasks. Similar transfer learning strategies can be useful for developing timely artificial intelligence solutions in future pandemic outbreaks.
ECG for high-throughput screening of multiple diseases: Proof-of-concept using multi-diagnosis deep learning from population-based datasets
Oct 06, 2022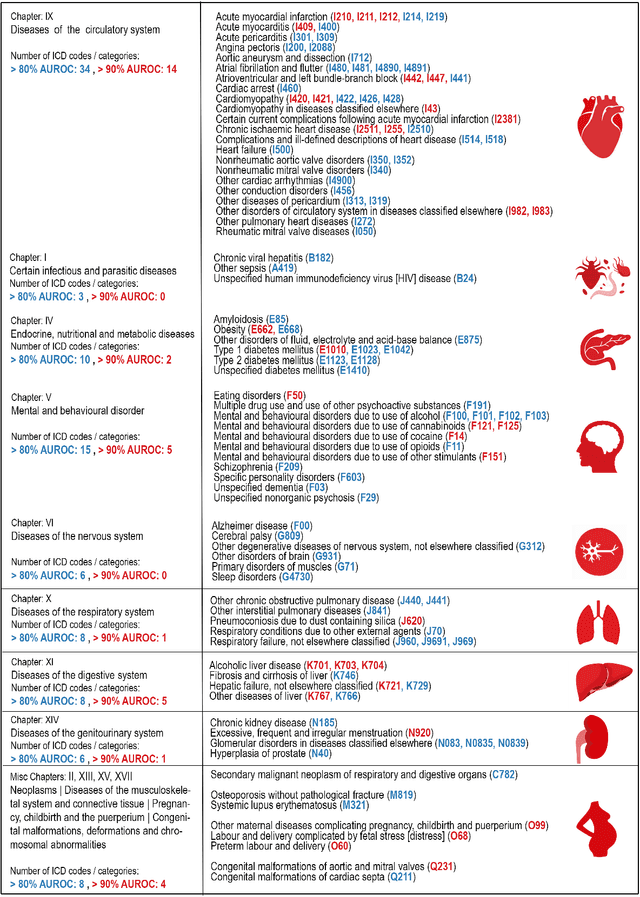
Electrocardiogram (ECG) abnormalities are linked to cardiovascular diseases, but may also occur in other non-cardiovascular conditions such as mental, neurological, metabolic and infectious conditions. However, most of the recent success of deep learning (DL) based diagnostic predictions in selected patient cohorts have been limited to a small set of cardiac diseases. In this study, we use a population-based dataset of >250,000 patients with >1000 medical conditions and >2 million ECGs to identify a wide range of diseases that could be accurately diagnosed from the patient's first in-hospital ECG. Our DL models uncovered 128 diseases and 68 disease categories with strong discriminative performance.
Domain-shift adaptation via linear transformations
Jan 14, 2022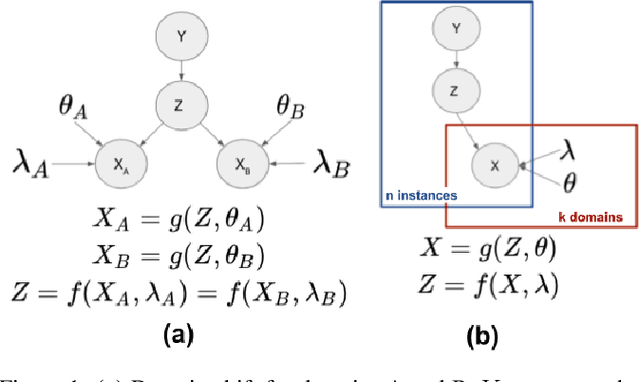
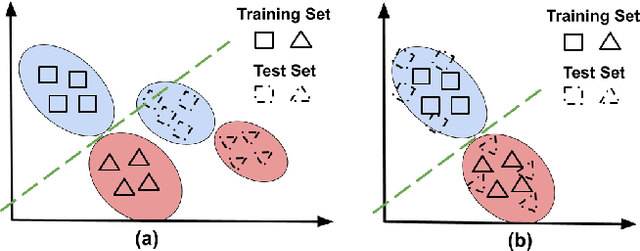
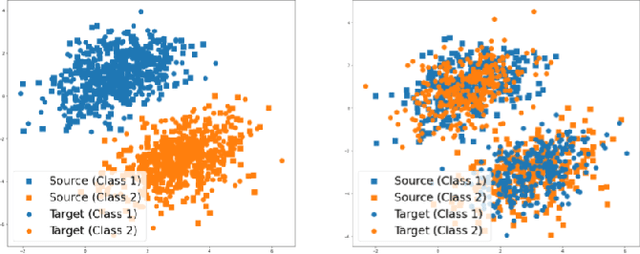
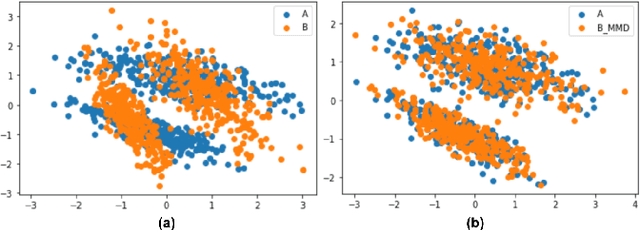
A predictor, $f_A : X \to Y$, learned with data from a source domain (A) might not be accurate on a target domain (B) when their distributions are different. Domain adaptation aims to reduce the negative effects of this distribution mismatch. Here, we analyze the case where $P_A(Y\ |\ X) \neq P_B(Y\ |\ X)$, $P_A(X) \neq P_B(X)$ but $P_A(Y) = P_B(Y)$; where there are affine transformations of $X$ that makes all distributions equivalent. We propose an approach to project the source and target domains into a lower-dimensional, common space, by (1) projecting the domains into the eigenvectors of the empirical covariance matrices of each domain, then (2) finding an orthogonal matrix that minimizes the maximum mean discrepancy between the projections of both domains. For arbitrary affine transformations, there is an inherent unidentifiability problem when performing unsupervised domain adaptation that can be alleviated in the semi-supervised case. We show the effectiveness of our approach in simulated data and in binary digit classification tasks, obtaining improvements up to 48% accuracy when correcting for the domain shift in the data.
Variational Auto-Encoder Architectures that Excel at Causal Inference
Nov 11, 2021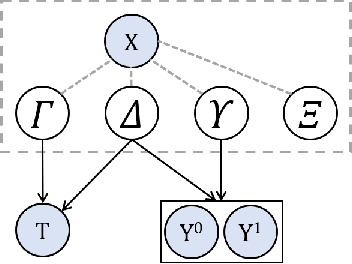
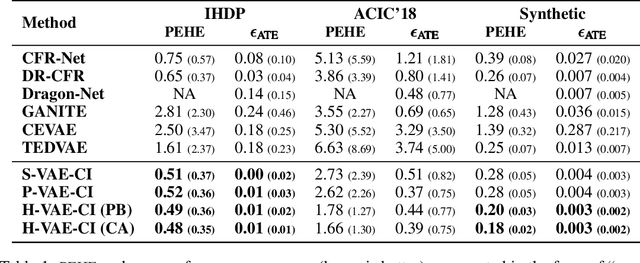
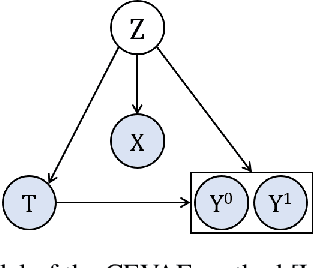

Estimating causal effects from observational data (at either an individual -- or a population -- level) is critical for making many types of decisions. One approach to address this task is to learn decomposed representations of the underlying factors of data; this becomes significantly more challenging when there are confounding factors (which influence both the cause and the effect). In this paper, we take a generative approach that builds on the recent advances in Variational Auto-Encoders to simultaneously learn those underlying factors as well as the causal effects. We propose a progressive sequence of models, where each improves over the previous one, culminating in the Hybrid model. Our empirical results demonstrate that the performance of all three proposed models are superior to both state-of-the-art discriminative as well as other generative approaches in the literature.
SIMLR: Machine Learning inside the SIR model for COVID-19 Forecasting
Jun 03, 2021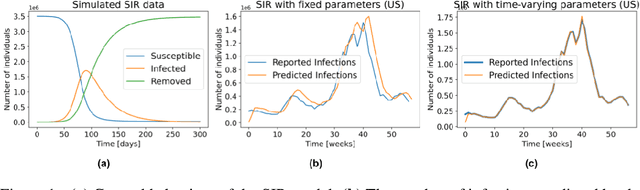
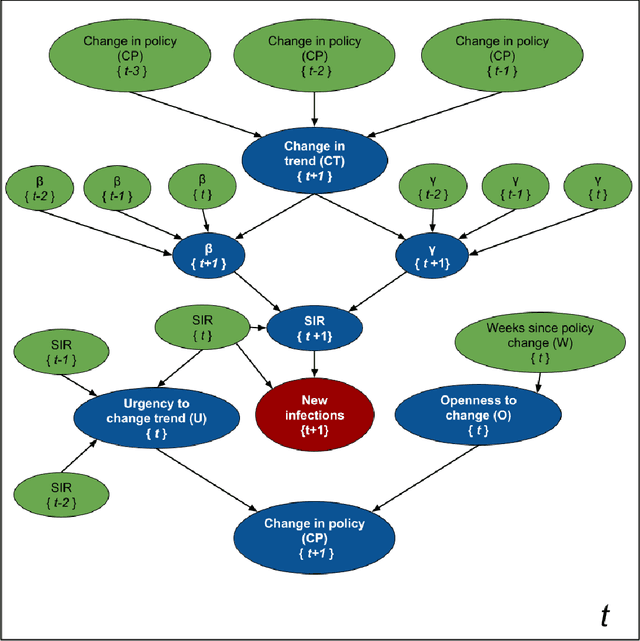
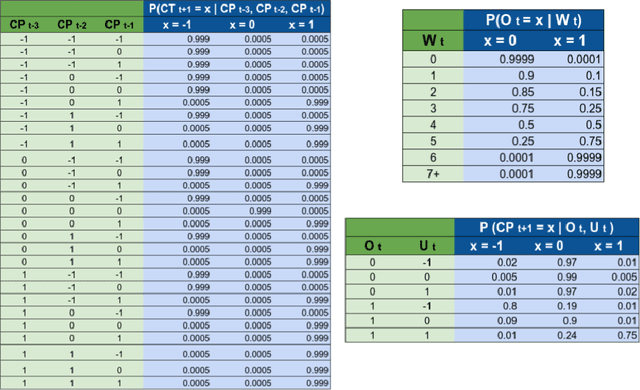
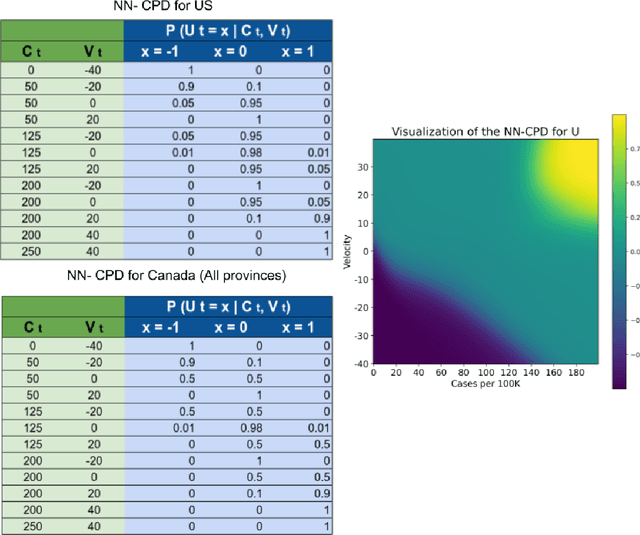
Accurate forecasts of the number of newly infected people during an epidemic are critical for making effective timely decisions. This paper addresses this challenge using the SIMLR model, which incorporates machine learning (ML) into the epidemiological SIR model. For each region, SIMLR tracks the changes in the policies implemented at the government level, which it uses to estimate the time-varying parameters of an SIR model for forecasting the number of new infections 1- to 4-weeks in advance.It also forecasts the probability of changes in those government policies at each of these future times, which is essential for the longer-range forecasts. We applied SIMLR to data from regions in Canada and in the United States,and show that its MAPE (mean average percentage error) performance is as good as SOTA forecasting models, with the added advantage of being an interpretable model. We expect that this approach will be useful not only for forecasting COVID-19 infections, but also in predicting the evolution of other infectious diseases.
Sample Efficient Learning of Image-Based Diagnostic Classifiers Using Probabilistic Labels
Feb 11, 2021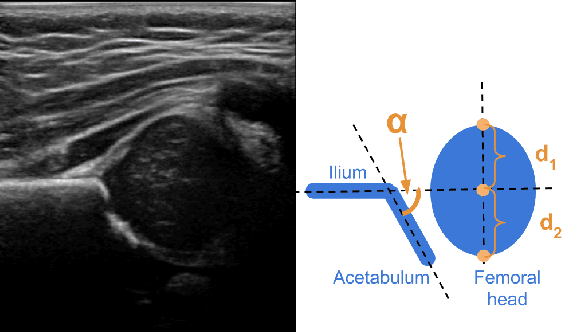
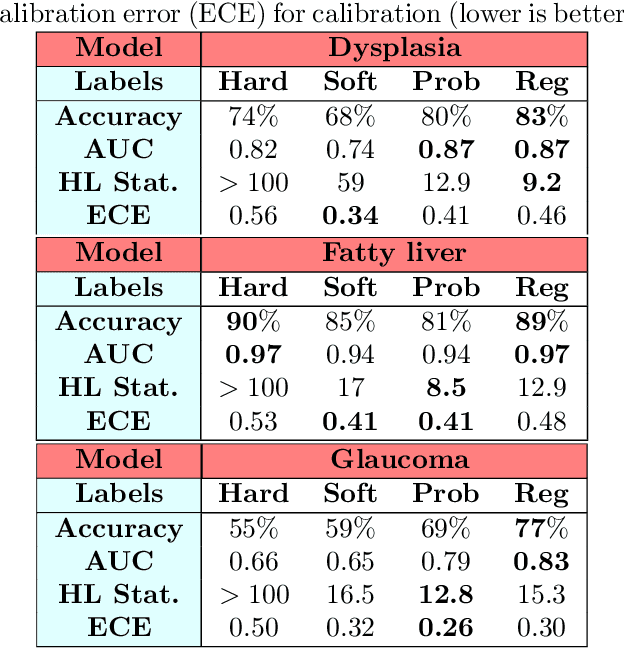
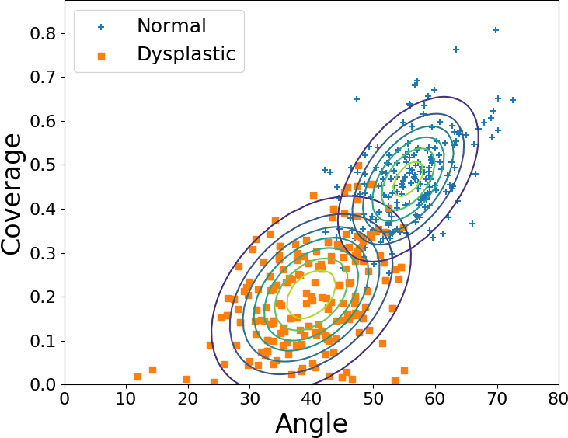
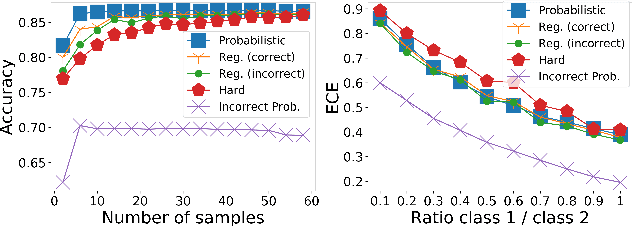
Deep learning approaches often require huge datasets to achieve good generalization. This complicates its use in tasks like image-based medical diagnosis, where the small training datasets are usually insufficient to learn appropriate data representations. For such sensitive tasks it is also important to provide the confidence in the predictions. Here, we propose a way to learn and use probabilistic labels to train accurate and calibrated deep networks from relatively small datasets. We observe gains of up to 22% in the accuracy of models trained with these labels, as compared with traditional approaches, in three classification tasks: diagnosis of hip dysplasia, fatty liver, and glaucoma. The outputs of models trained with probabilistic labels are calibrated, allowing the interpretation of its predictions as proper probabilities. We anticipate this approach will apply to other tasks where few training instances are available and expert knowledge can be encoded as probabilities.
Shared Space Transfer Learning for analyzing multi-site fMRI data
Oct 24, 2020
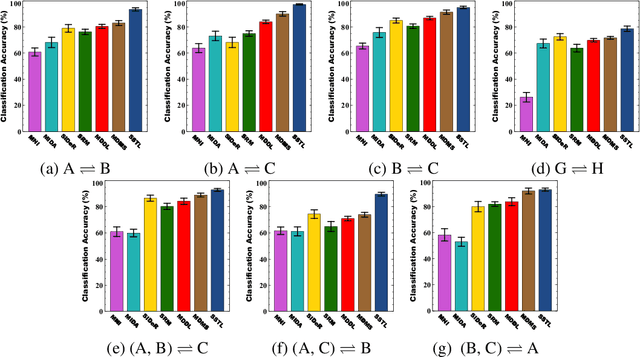
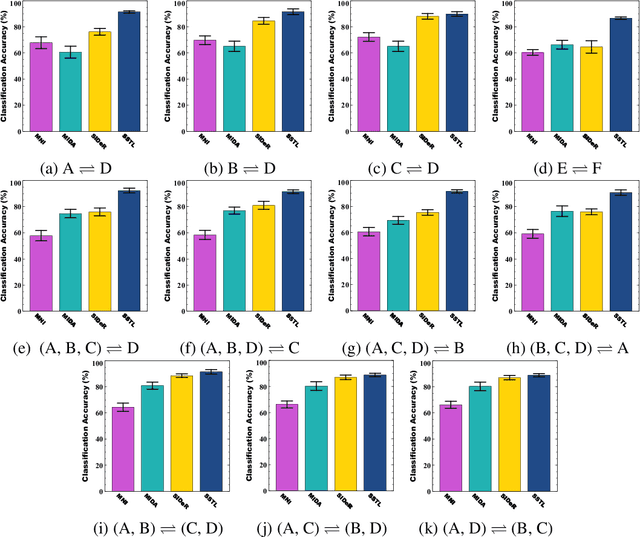
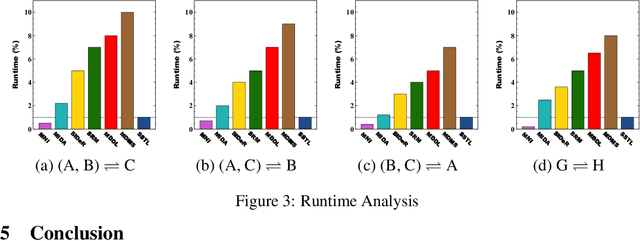
Multi-voxel pattern analysis (MVPA) learns predictive models from task-based functional magnetic resonance imaging (fMRI) data, for distinguishing when subjects are performing different cognitive tasks -- e.g., watching movies or making decisions. MVPA works best with a well-designed feature set and an adequate sample size. However, most fMRI datasets are noisy, high-dimensional, expensive to collect, and with small sample sizes. Further, training a robust, generalized predictive model that can analyze homogeneous cognitive tasks provided by multi-site fMRI datasets has additional challenges. This paper proposes the Shared Space Transfer Learning (SSTL) as a novel transfer learning (TL) approach that can functionally align homogeneous multi-site fMRI datasets, and so improve the prediction performance in every site. SSTL first extracts a set of common features for all subjects in each site. It then uses TL to map these site-specific features to a site-independent shared space in order to improve the performance of the MVPA. SSTL uses a scalable optimization procedure that works effectively for high-dimensional fMRI datasets. The optimization procedure extracts the common features for each site by using a single-iteration algorithm and maps these site-specific common features to the site-independent shared space. We evaluate the effectiveness of the proposed method for transferring between various cognitive tasks. Our comprehensive experiments validate that SSTL achieves superior performance to other state-of-the-art analysis techniques.