 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeEnhancing Target-Guided Proactive Dialogue Systems via Conversational Scenario Modeling and Intent-Keyword Bridging
May 12, 2026A target-guided proactive dialogue system aims to steer conversations proactively toward pre-defined targets, such as designated keywords or specific topics. During guided conversations, dynamically modeling conversational scenarios and intent keywords to guide system utterance generation is beneficial; however, existing work largely overlooks this aspect, resulting in a mismatch with the dynamics of real-world conversations. In this paper, we jointly model user profiles and domain knowledge as conversational scenarios to introduce a scenario bias that dynamically influences system utterances, and employ intent-keyword bridging to predict intent keywords for upcoming dialogue turns, providing higher level and more flexible guidance. Extensive automatic and human evaluations demonstrate the effectiveness of conversational scenario modeling and intent keyword bridging, yielding substantial improvements in proactivity, fluency, and informativeness for target-guided proactive dialogue systems, thereby narrowing the gap with real world interactions.
Multi-Hop Question Generation via Dual-Perspective Keyword Guidance
May 21, 2025Multi-hop question generation (MQG) aims to generate questions that require synthesizing multiple information snippets from documents to derive target answers. The primary challenge lies in effectively pinpointing crucial information snippets related to question-answer (QA) pairs, typically relying on keywords. However, existing works fail to fully utilize the guiding potential of keywords and neglect to differentiate the distinct roles of question-specific and document-specific keywords. To address this, we define dual-perspective keywords (i.e., question and document keywords) and propose a Dual-Perspective Keyword-Guided (DPKG) framework, which seamlessly integrates keywords into the multi-hop question generation process. We argue that question keywords capture the questioner's intent, whereas document keywords reflect the content related to the QA pair. Functionally, question and document keywords work together to pinpoint essential information snippets in the document, with question keywords required to appear in the generated question. The DPKG framework consists of an expanded transformer encoder and two answer-aware transformer decoders for keyword and question generation, respectively. Extensive experiments demonstrate the effectiveness of our work, showcasing its promising performance and underscoring its significant value in the MQG task.
Enhanced Sampling, Public Dataset and Generative Model for Drug-Protein Dissociation Dynamics
Apr 25, 2025Drug-protein binding and dissociation dynamics are fundamental to understanding molecular interactions in biological systems. While many tools for drug-protein interaction studies have emerged, especially artificial intelligence (AI)-based generative models, predictive tools on binding/dissociation kinetics and dynamics are still limited. We propose a novel research paradigm that combines molecular dynamics (MD) simulations, enhanced sampling, and AI generative models to address this issue. We propose an enhanced sampling strategy to efficiently implement the drug-protein dissociation process in MD simulations and estimate the free energy surface (FES). We constructed a program pipeline of MD simulations based on this sampling strategy, thus generating a dataset including 26,612 drug-protein dissociation trajectories containing about 13 million frames. We named this dissociation dynamics dataset DD-13M and used it to train a deep equivariant generative model UnbindingFlow, which can generate collision-free dissociation trajectories. The DD-13M database and UnbindingFlow model represent a significant advancement in computational structural biology, and we anticipate its broad applicability in machine learning studies of drug-protein interactions. Our ongoing efforts focus on expanding this methodology to encompass a broader spectrum of drug-protein complexes and exploring novel applications in pathway prediction.
Deep Reinforcement Learning of Transition States
Nov 13, 2020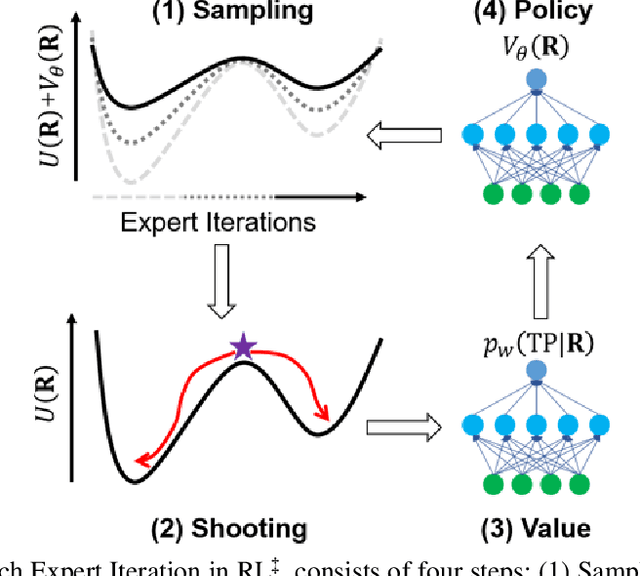
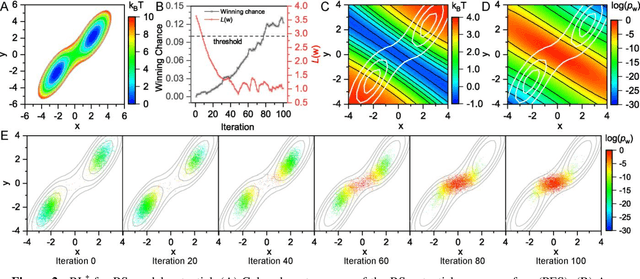
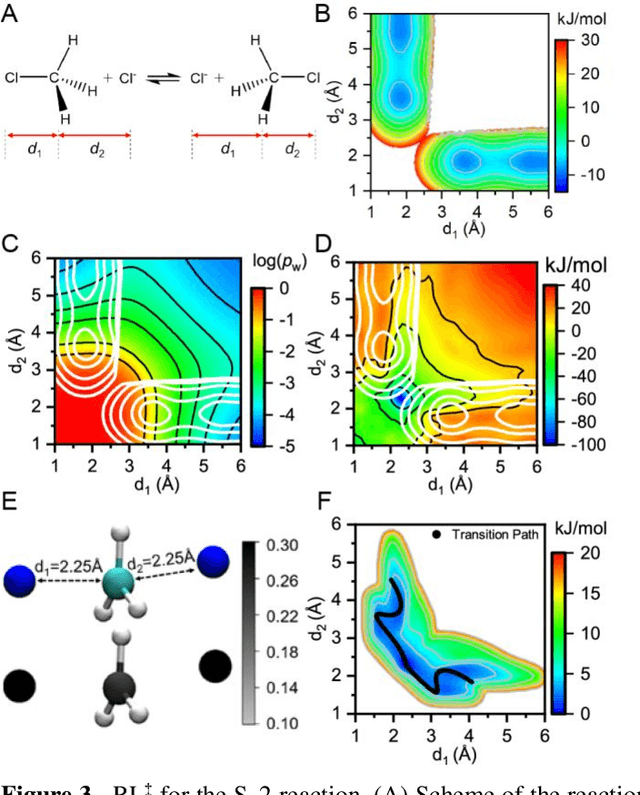
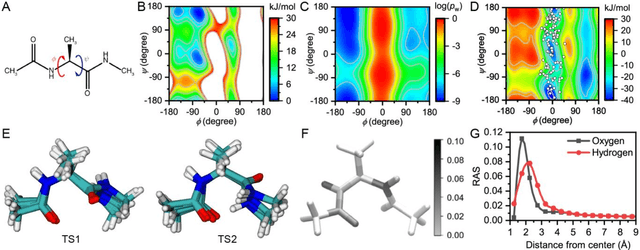
Combining reinforcement learning (RL) and molecular dynamics (MD) simulations, we propose a machine-learning approach (RL$^\ddag$) to automatically unravel chemical reaction mechanisms. In RL$^\ddag$, locating the transition state of a chemical reaction is formulated as a game, where a virtual player is trained to shoot simulation trajectories connecting the reactant and product. The player utilizes two functions, one for value estimation and the other for policy making, to iteratively improve the chance of winning this game. We can directly interpret the reaction mechanism according to the value function. Meanwhile, the policy function enables efficient sampling of the transition paths, which can be further used to analyze the reaction dynamics and kinetics. Through multiple experiments, we show that RL{\ddag} can be trained tabula rasa hence allows us to reveal chemical reaction mechanisms with minimal subjective biases.
A Perspective on Deep Learning for Molecular Modeling and Simulations
Apr 25, 2020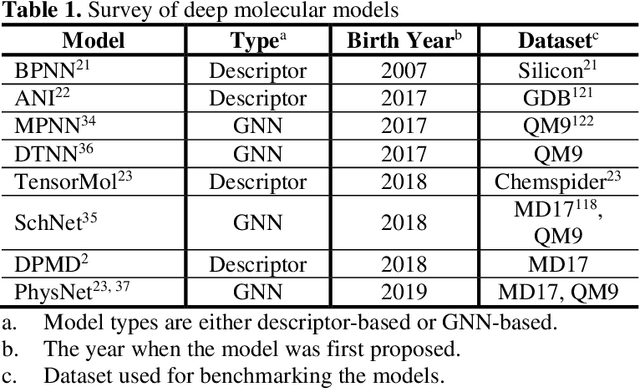
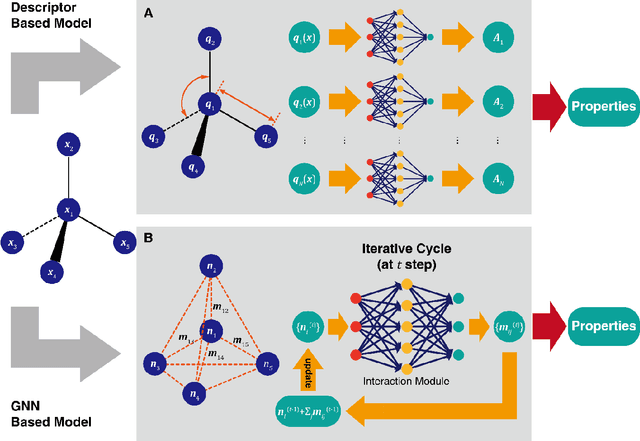
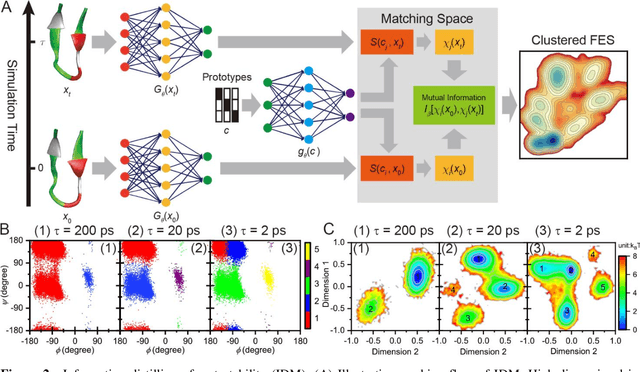
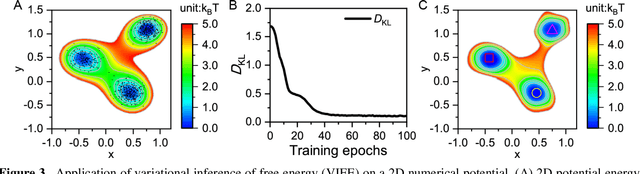
Deep learning is transforming many areas in science, and it has great potential in modeling molecular systems. However, unlike the mature deployment of deep learning in computer vision and natural language processing, its development in molecular modeling and simulations is still at an early stage, largely because the inductive biases of molecules are completely different from those of images or texts. Footed on these differences, we first reviewed the limitations of traditional deep learning models from the perspective of molecular physics, and wrapped up some relevant technical advancement at the interface between molecular modeling and deep learning. We do not focus merely on the ever more complex neural network models, instead, we emphasize the theories and ideas behind modern deep learning. We hope that transacting these ideas into molecular modeling will create new opportunities. For this purpose, we summarized several representative applications, ranging from supervised to unsupervised and reinforcement learning, and discussed their connections with the emerging trends in deep learning. Finally, we outlook promising directions which may help address the existing issues in the current framework of deep molecular modeling.