 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGrading Scale Impact on LLM-as-a-Judge: Human-LLM Alignment Is Highest on 0-5 Grading Scale
Jan 06, 2026Large language models (LLMs) are increasingly used as automated evaluators, yet prior works demonstrate that these LLM judges often lack consistency in scoring when the prompt is altered. However, the effect of the grading scale itself remains underexplored. We study the LLM-as-a-judge problem by comparing two kinds of raters: humans and LLMs. We collect ratings from both groups on three scales and across six benchmarks that include objective, open-ended subjective, and mixed tasks. Using intraclass correlation coefficients (ICC) to measure absolute agreement, we find that LLM judgments are not perfectly consistent across scales on subjective benchmarks, and that the choice of scale substantially shifts human-LLM agreement, even when within-group panel reliability is high. Aggregated over tasks, the grading scale of 0-5 yields the strongest human-LLM alignment. We further demonstrate that pooled reliability can mask benchmark heterogeneity and reveal systematic subgroup differences in alignment across gender groups, strengthening the importance of scale design and sub-level diagnostics as essential components of LLM-as-a-judge protocols.
Automated Model-Free Sorting of Single-Molecule Fluorescence Events Using a Deep Learning Based Hidden-State Model
May 13, 2025Single-molecule fluorescence assays enable high-resolution analysis of biomolecular dynamics, but traditional analysis pipelines are labor-intensive and rely on users' experience, limiting scalability and reproducibility. Recent deep learning models have automated aspects of data processing, yet many still require manual thresholds, complex architectures, or extensive labeled data. Therefore, we present DASH, a fully streamlined architecture for trace classification, state assignment, and automatic sorting that requires no user input. DASH demonstrates robust performance across users and experimental conditions both in equilibrium and non-equilibrium systems such as Cas12a-mediated DNA cleavage. This paper proposes a novel strategy for the automatic and detailed sorting of single-molecule fluorescence events. The dynamic cleavage process of Cas12a is used as an example to provide a comprehensive analysis. This approach is crucial for studying biokinetic structural changes at the single-molecule level.
MM-Skin: Enhancing Dermatology Vision-Language Model with an Image-Text Dataset Derived from Textbooks
May 09, 2025Medical vision-language models (VLMs) have shown promise as clinical assistants across various medical fields. However, specialized dermatology VLM capable of delivering professional and detailed diagnostic analysis remains underdeveloped, primarily due to less specialized text descriptions in current dermatology multimodal datasets. To address this issue, we propose MM-Skin, the first large-scale multimodal dermatology dataset that encompasses 3 imaging modalities, including clinical, dermoscopic, and pathological and nearly 10k high-quality image-text pairs collected from professional textbooks. In addition, we generate over 27k diverse, instruction-following vision question answering (VQA) samples (9 times the size of current largest dermatology VQA dataset). Leveraging public datasets and MM-Skin, we developed SkinVL, a dermatology-specific VLM designed for precise and nuanced skin disease interpretation. Comprehensive benchmark evaluations of SkinVL on VQA, supervised fine-tuning (SFT) and zero-shot classification tasks across 8 datasets, reveal its exceptional performance for skin diseases in comparison to both general and medical VLM models. The introduction of MM-Skin and SkinVL offers a meaningful contribution to advancing the development of clinical dermatology VLM assistants. MM-Skin is available at https://github.com/ZwQ803/MM-Skin
Enhanced Sampling, Public Dataset and Generative Model for Drug-Protein Dissociation Dynamics
Apr 25, 2025Drug-protein binding and dissociation dynamics are fundamental to understanding molecular interactions in biological systems. While many tools for drug-protein interaction studies have emerged, especially artificial intelligence (AI)-based generative models, predictive tools on binding/dissociation kinetics and dynamics are still limited. We propose a novel research paradigm that combines molecular dynamics (MD) simulations, enhanced sampling, and AI generative models to address this issue. We propose an enhanced sampling strategy to efficiently implement the drug-protein dissociation process in MD simulations and estimate the free energy surface (FES). We constructed a program pipeline of MD simulations based on this sampling strategy, thus generating a dataset including 26,612 drug-protein dissociation trajectories containing about 13 million frames. We named this dissociation dynamics dataset DD-13M and used it to train a deep equivariant generative model UnbindingFlow, which can generate collision-free dissociation trajectories. The DD-13M database and UnbindingFlow model represent a significant advancement in computational structural biology, and we anticipate its broad applicability in machine learning studies of drug-protein interactions. Our ongoing efforts focus on expanding this methodology to encompass a broader spectrum of drug-protein complexes and exploring novel applications in pathway prediction.
A Note on Learning Rare Events in Molecular Dynamics using LSTM and Transformer
Jul 14, 2021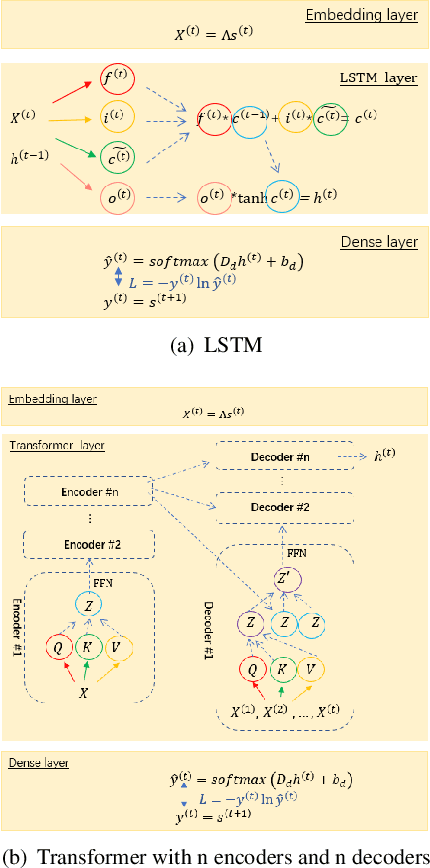
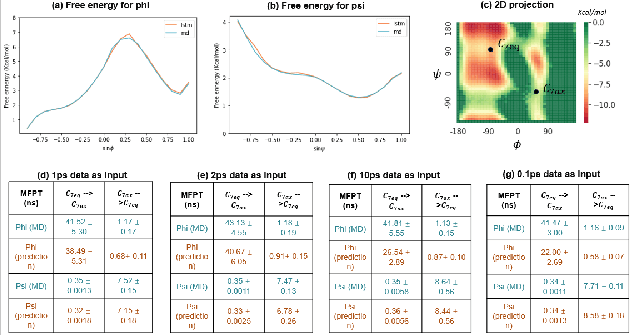
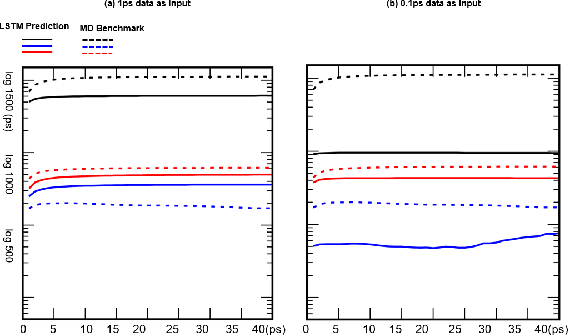
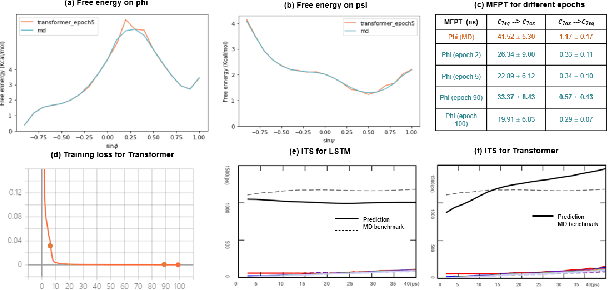
Recurrent neural networks for language models like long short-term memory (LSTM) have been utilized as a tool for modeling and predicting long term dynamics of complex stochastic molecular systems. Recently successful examples on learning slow dynamics by LSTM are given with simulation data of low dimensional reaction coordinate. However, in this report we show that the following three key factors significantly affect the performance of language model learning, namely dimensionality of reaction coordinates, temporal resolution and state partition. When applying recurrent neural networks to molecular dynamics simulation trajectories of high dimensionality, we find that rare events corresponding to the slow dynamics might be obscured by other faster dynamics of the system, and cannot be efficiently learned. Under such conditions, we find that coarse graining the conformational space into metastable states and removing recrossing events when estimating transition probabilities between states could greatly help improve the accuracy of slow dynamics learning in molecular dynamics. Moreover, we also explore other models like Transformer, which do not show superior performance than LSTM in overcoming these issues. Therefore, to learn rare events of slow molecular dynamics by LSTM and Transformer, it is critical to choose proper temporal resolution (i.e., saving intervals of MD simulation trajectories) and state partition in high resolution data, since deep neural network models might not automatically disentangle slow dynamics from fast dynamics when both are present in data influencing each other.