 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMaterialsAtlas.org: A Materials Informatics Web App Platform for Materials Discovery and Survey of State-of-the-Art
Sep 09, 2021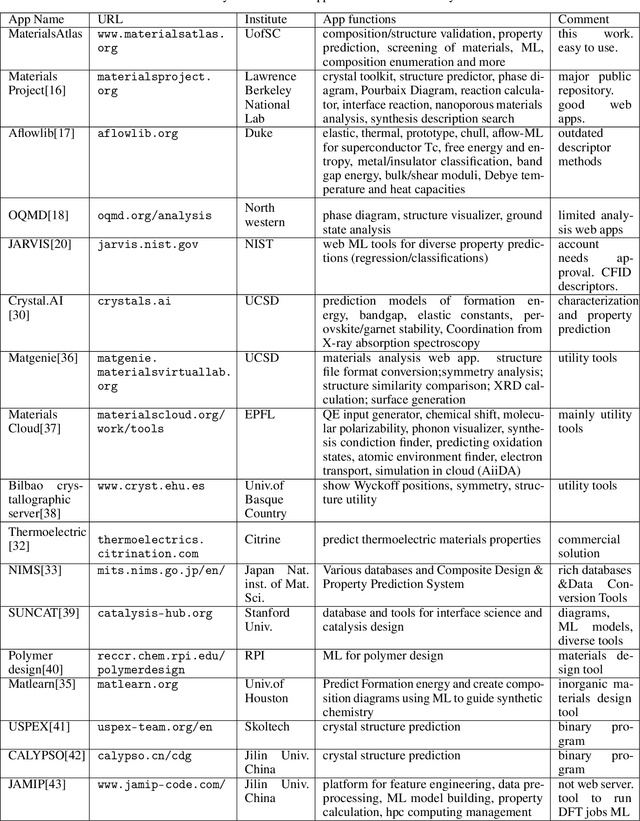
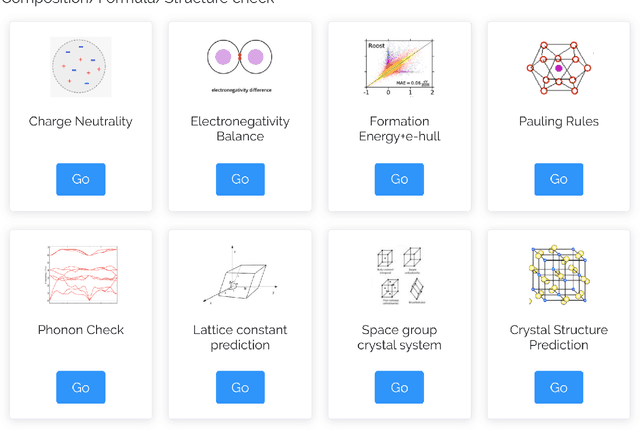
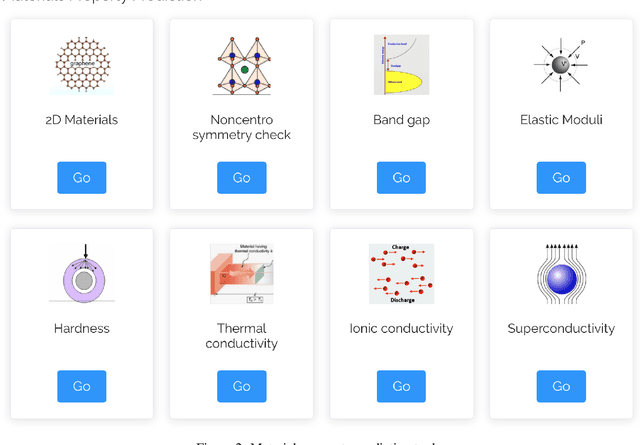
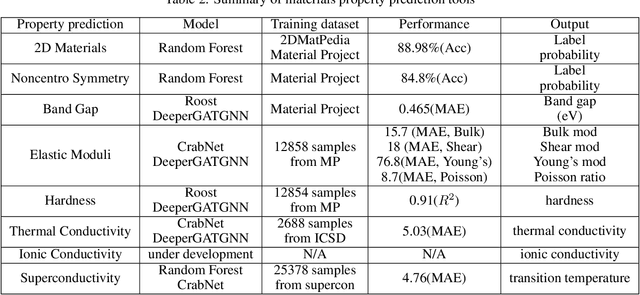
The availability and easy access of large scale experimental and computational materials data have enabled the emergence of accelerated development of algorithms and models for materials property prediction, structure prediction, and generative design of materials. However, lack of user-friendly materials informatics web servers has severely constrained the wide adoption of such tools in the daily practice of materials screening, tinkering, and design space exploration by materials scientists. Herein we first survey current materials informatics web apps and then propose and develop MaterialsAtlas.org, a web based materials informatics toolbox for materials discovery, which includes a variety of routinely needed tools for exploratory materials discovery, including materials composition and structure check (e.g. for neutrality, electronegativity balance, dynamic stability, Pauling rules), materials property prediction (e.g. band gap, elastic moduli, hardness, thermal conductivity), and search for hypothetical materials. These user-friendly tools can be freely accessed at \url{www.materialsatlas.org}. We argue that such materials informatics apps should be widely developed by the community to speed up the materials discovery processes.
Crystal structure prediction of materials with high symmetry using differential evolution
Apr 20, 2021
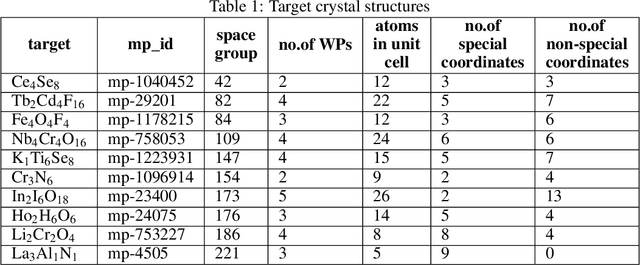
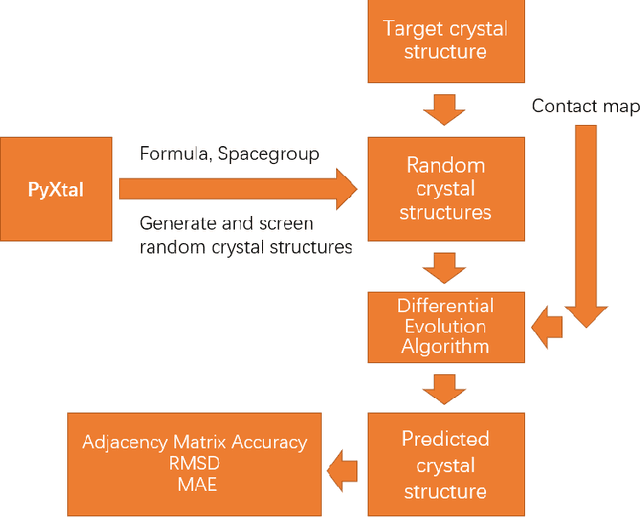
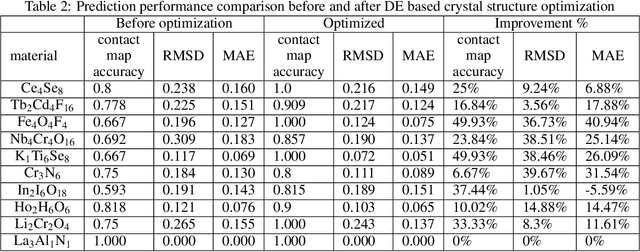
Crystal structure determines properties of materials. With the crystal structure of a chemical substance, many physical and chemical properties can be predicted by first-principles calculations or machine learning models. Since it is relatively easy to generate a hypothetical chemically valid formula, crystal structure prediction becomes an important method for discovering new materials. In our previous work, we proposed a contact map-based crystal structure prediction method, which uses global optimization algorithms such as genetic algorithms to maximize the match between the contact map of the predicted structure and the contact map of the real crystal structure to search for the coordinates at the Wyckoff Positions(WP). However, when predicting the crystal structure with high symmetry, we found that the global optimization algorithm has difficulty to find an effective combination of WPs that satisfies the chemical formula, which is mainly caused by the inconsistency between the dimensionality of the contact map of the predicted crystal structure and the dimensionality of the contact map of the target crystal structure. This makes it challenging to predict the crystal structures of high-symmetry crystals. In order to solve this problem, here we propose to use PyXtal to generate and filter random crystal structures with given symmetry constraints based on the information such as chemical formulas and space groups. With contact map as the optimization goal, we use differential evolution algorithms to search for non-special coordinates at the Wyckoff positions to realize the structure prediction of high-symmetry crystal materials. Our experimental results show that our proposed algorithm CMCrystalHS can effectively solve the problem of inconsistent contact map dimensions and predict the crystal structures with high symmetry.
SoundCLR: Contrastive Learning of Representations For Improved Environmental Sound Classification
Mar 02, 2021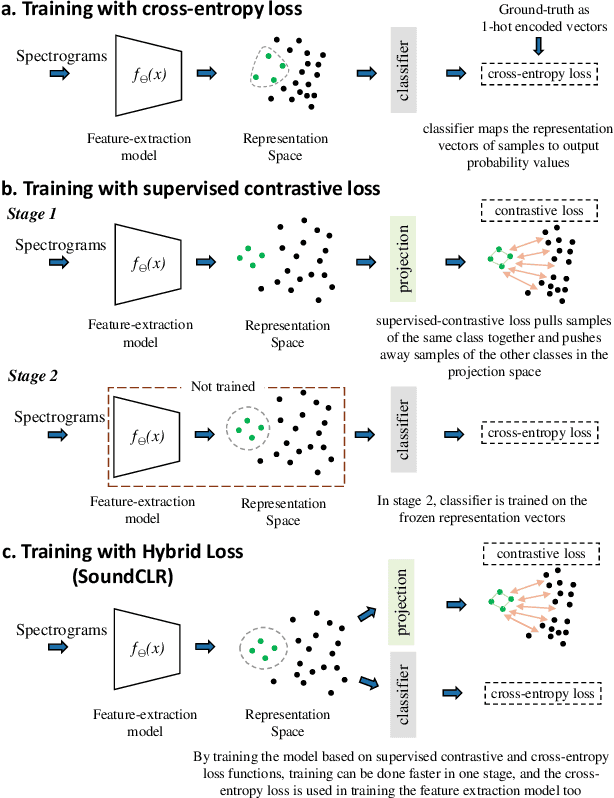
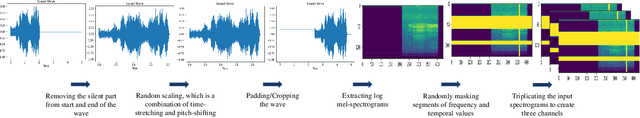
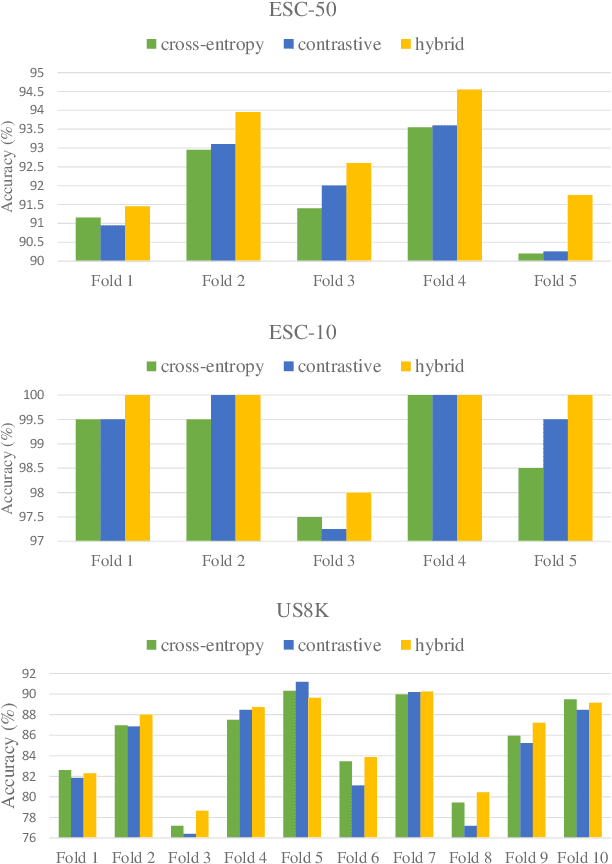
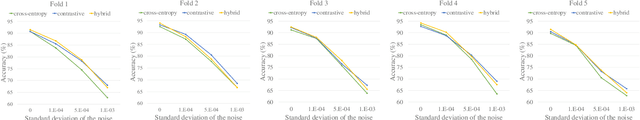
Environmental Sound Classification (ESC) is a challenging field of research in non-speech audio processing. Most of current research in ESC focuses on designing deep models with special architectures tailored for specific audio datasets, which usually cannot exploit the intrinsic patterns in the data. However recent studies have surprisingly shown that transfer learning from models trained on ImageNet is a very effective technique in ESC. Herein, we propose SoundCLR, a supervised contrastive learning method for effective environment sound classification with state-of-the-art performance, which works by learning representations that disentangle the samples of each class from those of other classes. Our deep network models are trained by combining a contrastive loss that contributes to a better probability output by the classification layer with a cross-entropy loss on the output of the classifier layer to map the samples to their respective 1-hot encoded labels. Due to the comparatively small sizes of the available environmental sound datasets, we propose and exploit a transfer learning and strong data augmentation pipeline and apply the augmentations on both the sound signals and their log-mel spectrograms before inputting them to the model. Our experiments show that our masking based augmentation technique on the log-mel spectrograms can significantly improve the recognition performance. Our extensive benchmark experiments show that our hybrid deep network models trained with combined contrastive and cross-entropy loss achieved the state-of-the-art performance on three benchmark datasets ESC-10, ESC-50, and US8K with validation accuracies of 99.75\%, 93.4\%, and 86.49\% respectively. The ensemble version of our models also outperforms other top ensemble methods. The code is available at https://github.com/alireza-nasiri/SoundCLR.
Active learning based generative design for the discovery of wide bandgap materials
Feb 28, 2021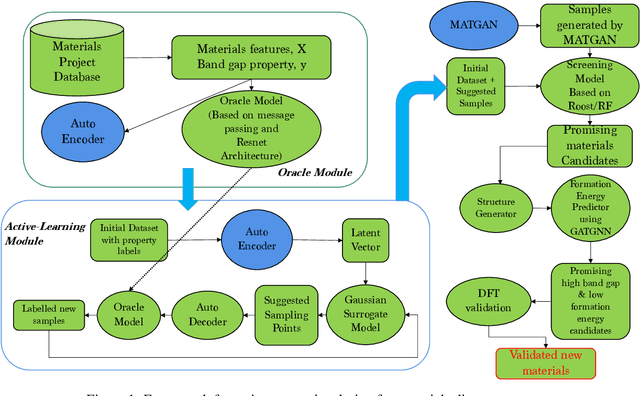
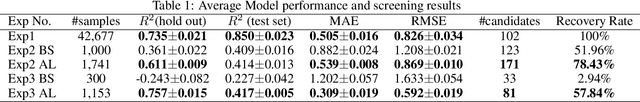
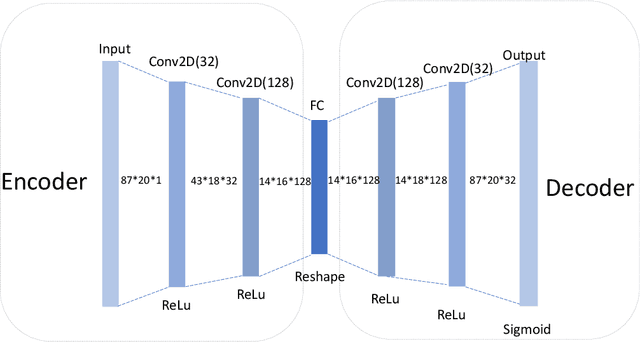
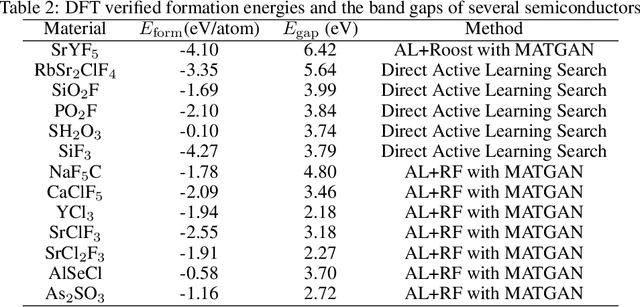
Active learning has been increasingly applied to screening functional materials from existing materials databases with desired properties. However, the number of known materials deposited in the popular materials databases such as ICSD and Materials Project is extremely limited and consists of just a tiny portion of the vast chemical design space. Herein we present an active generative inverse design method that combines active learning with a deep variational autoencoder neural network and a generative adversarial deep neural network model to discover new materials with a target property in the whole chemical design space. The application of this method has allowed us to discover new thermodynamically stable materials with high band gap (SrYF$_5$) and semiconductors with specified band gap ranges (SrClF$_3$, CaClF$_5$, YCl$_3$, SrC$_2$F$_3$, AlSCl, As$_2$O$_3$), all of which are verified by the first principle DFT calculations. Our experiments show that while active learning itself may sample chemically infeasible candidates, these samples help to train effective screening models for filtering out materials with desired properties from the hypothetical materials created by the generative model. The experiments show the effectiveness of our active generative inverse design approach.
NODE-SELECT: A Graph Neural Network Based On A Selective Propagation Technique
Feb 17, 2021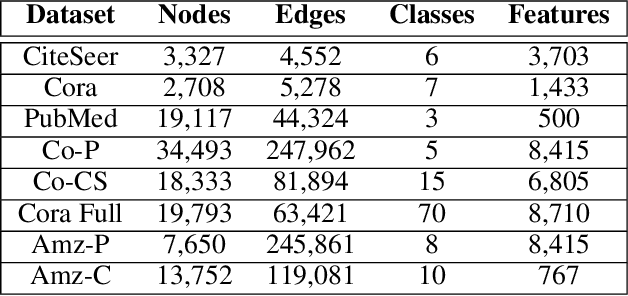
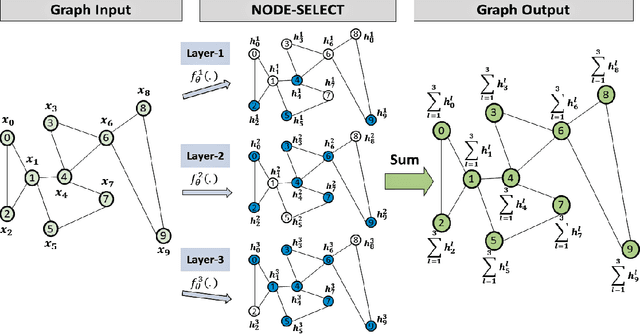
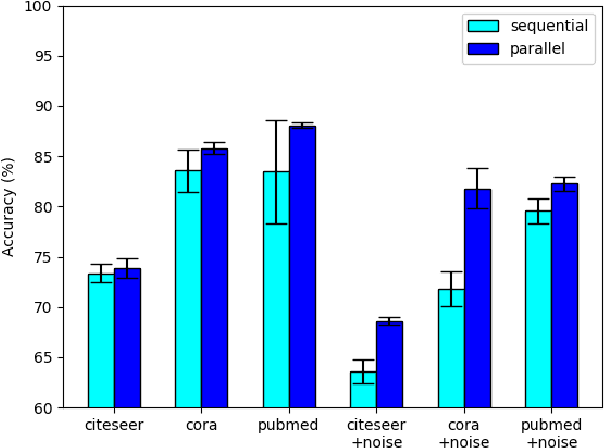
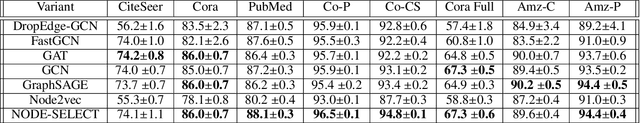
While there exists a wide variety of graph neural networks (GNN) for node classification, only a minority of them adopt mechanisms that effectively target noise propagation during the message-passing procedure. Additionally, a very important challenge that significantly affects graph neural networks is the issue of scalability which limits their application to larger graphs. In this paper we propose our method named NODE-SELECT: an efficient graph neural network that uses subsetting layers which only allow the best sharing-fitting nodes to propagate their information. By having a selection mechanism within each layer which we stack in parallel, our proposed method NODE-SELECT is able to both reduce the amount noise propagated and adapt the restrictive sharing concept observed in real world graphs. Our NODE-SELECT significantly outperformed existing GNN frameworks in noise experiments and matched state-of-the art results in experiments without noise over different benchmark datasets.
Computational discovery of new 2D materials using deep learning generative models
Dec 16, 2020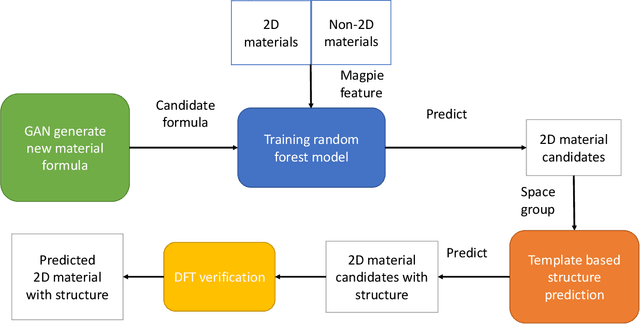
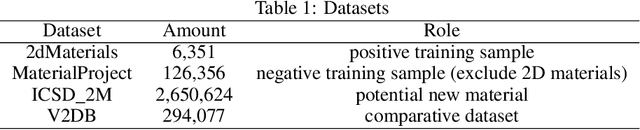
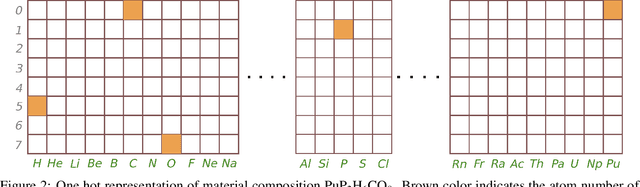
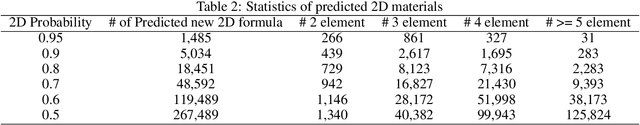
Two dimensional (2D) materials have emerged as promising functional materials with many applications such as semiconductors and photovoltaics because of their unique optoelectronic properties. While several thousand 2D materials have been screened in existing materials databases, discovering new 2D materials remains to be challenging. Herein we propose a deep learning generative model for composition generation combined with random forest based 2D materials classifier to discover new hypothetical 2D materials. Furthermore, a template based element substitution structure prediction approach is developed to predict the crystal structures of a subset of the newly predicted hypothetical formulas, which allows us to confirm their structure stability using DFT calculations. So far, we have discovered 267,489 new potential 2D materials compositions and confirmed twelve 2D/layered materials by DFT formation energy calculation. Our results show that generative machine learning models provide an effective way to explore the vast chemical design space for new 2D materials discovery.
A Survey on Machine Reading Comprehension: Tasks, Evaluation Metrics, and Benchmark Datasets
Jun 21, 2020

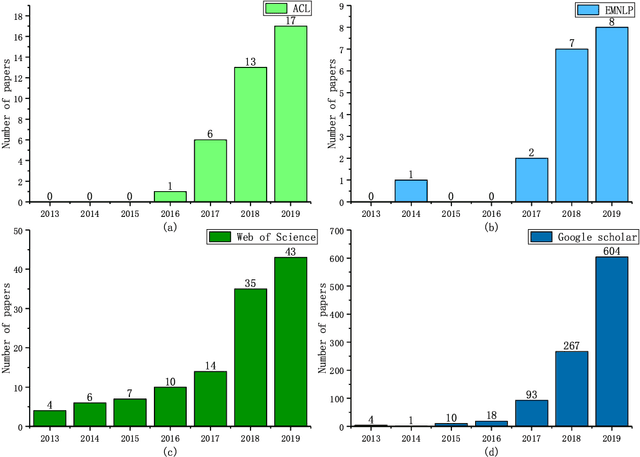
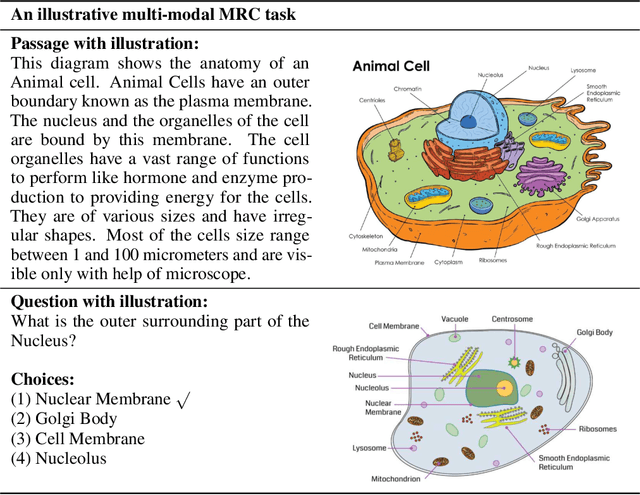
Machine Reading Comprehension (MRC) is a challenging NLP research field with wide real world applications. The great progress of this field in recent years is mainly due to the emergence of large-scale datasets and deep learning. At present, a lot of MRC models have already surpassed the human performance on many datasets despite the obvious giant gap between existing MRC models and genuine human-level reading comprehension. This shows the need of improving existing datasets, evaluation metrics and models to move the MRC models toward 'real' understanding. To address this lack of comprehensive survey of existing MRC tasks, evaluation metrics and datasets, herein, (1) we analyzed 57 MRC tasks and datasets; proposed a more precise classification method of MRC tasks with 4 different attributes (2) we summarized 9 evaluation metrics of MRC tasks and (3) 7 attributes and 10 characteristics of MRC datasets; (4) We also discussed some open issues in MRC research and highlight some future research directions. In addition, to help the community, we have collected, organized, and published our data on a companion website(https://mrc-datasets.github.io/) where MRC researchers could directly access each MRC dataset, papers, baseline projects and browse the leaderboard.
Predicting Elastic Properties of Materials from Electronic Charge Density Using 3D Deep Convolutional Neural Networks
Apr 11, 2020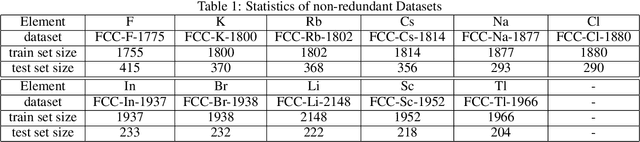
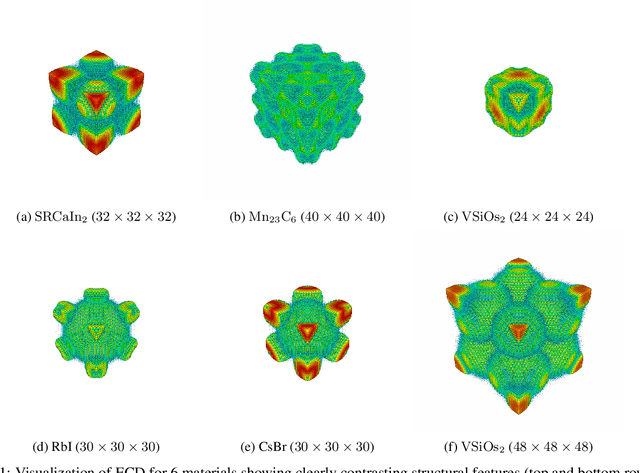
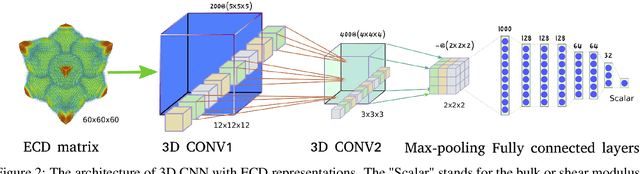

Materials representation plays a key role in machine learning based prediction of materials properties and new materials discovery. Currently both graph and 3D voxel representation methods are based on the heterogeneous elements of the crystal structures. Here, we propose to use electronic charge density (ECD) as a generic unified 3D descriptor for materials property prediction with the advantage of possessing close relation with the physical and chemical properties of materials. We developed an ECD based 3D convolutional neural networks (CNNs) for predicting elastic properties of materials, in which CNNs can learn effective hierarchical features with multiple convolving and pooling operations. Extensive benchmark experiments over 2,170 Fm-3m face-centered-cubic (FCC) materials show that our ECD based CNNs can achieve good performance for elasticity prediction. Especially, our CNN models based on the fusion of elemental Magpie features and ECD descriptors achieved the best 5-fold cross-validation performance. More importantly, we showed that our ECD based CNN models can achieve significantly better extrapolation performance when evaluated over non-redundant datasets where there are few neighbor training samples around test samples. As additional validation, we evaluated the predictive performance of our models on 329 materials of space group Fm-3m by comparing to DFT calculated values, which shows better prediction power of our model for bulk modulus than shear modulus. Due to the unified representation power of ECD, it is expected that our ECD based CNN approach can also be applied to predict other physical and chemical properties of crystalline materials.
Global Attention based Graph Convolutional Neural Networks for Improved Materials Property Prediction
Mar 11, 2020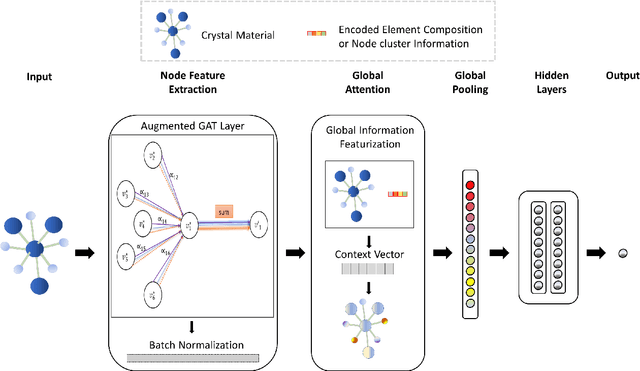

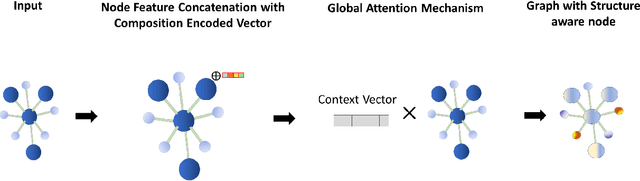
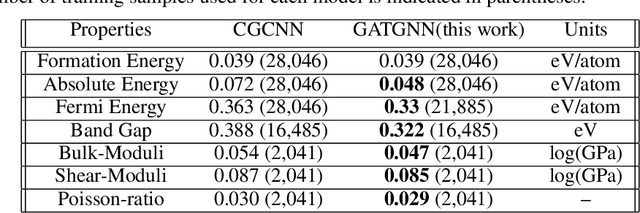
Machine learning (ML) methods have gained increasing popularity in exploring and developing new materials. More specifically, graph neural network (GNN) has been applied in predicting material properties. In this work, we develop a novel model, GATGNN, for predicting inorganic material properties based on graph neural networks composed of multiple graph-attention layers (GAT) and a global attention layer. Through the application of the GAT layers, our model can efficiently learn the complex bonds shared among the atoms within each atom's local neighborhood. Subsequently, the global attention layer provides the weight coefficients of each atom in the inorganic crystal material which are used to considerably improve our model's performance. Notably, with the development of our GATGNN model, we show that our method is able to both outperform the previous models' predictions and provide insight into the crystallization of the material.
Machine Learning based prediction of noncentrosymmetric crystal materials
Feb 26, 2020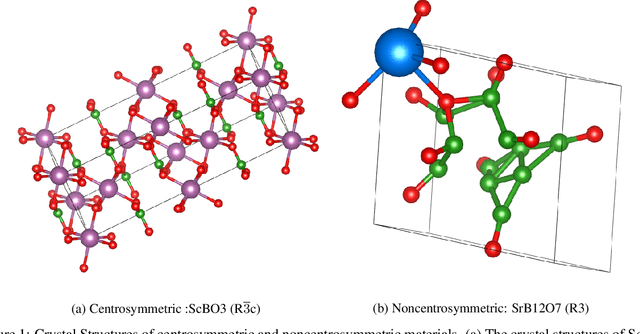
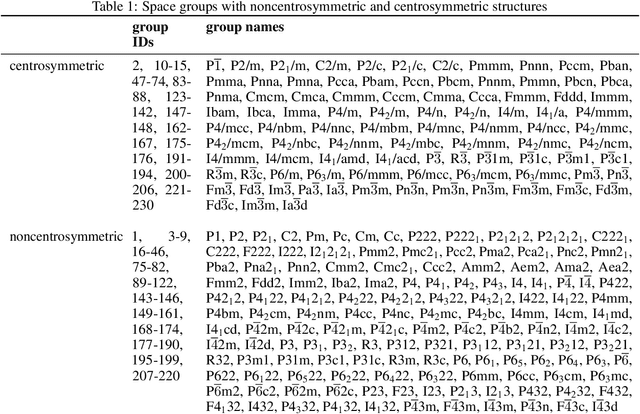
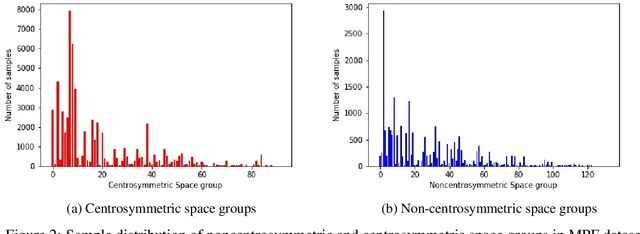
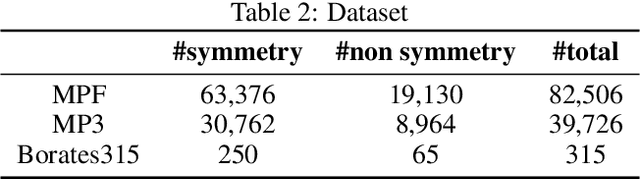
Noncentrosymmetric materials play a critical role in many important applications such as laser technology, communication systems,quantum computing, cybersecurity, and etc. However, the experimental discovery of new noncentrosymmetric materials is extremely difficult. Here we present a machine learning model that could predict whether the composition of a potential crystalline structure would be centrosymmetric or not. By evaluating a diverse set of composition features calculated using matminer featurizer package coupled with different machine learning algorithms, we find that Random Forest Classifiers give the best performance for noncentrosymmetric material prediction, reaching an accuracy of 84.8% when evaluated with 10 fold cross-validation on the dataset with 82,506 samples extracted from Materials Project. A random forest model trained with materials with only 3 elements gives even higher accuracy of 86.9%. We apply our ML model to screen potential noncentrosymmetric materials from 2,000,000 hypothetical materials generated by our inverse design engine and report the top 20 candidate noncentrosymmetric materials with 2 to 4 elements and top 20 borate candidates