 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMachine Learning based prediction of noncentrosymmetric crystal materials
Feb 26, 2020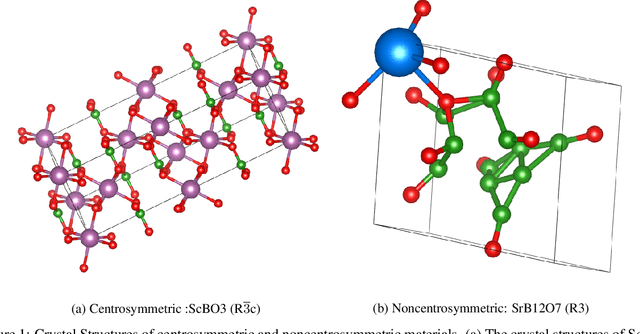
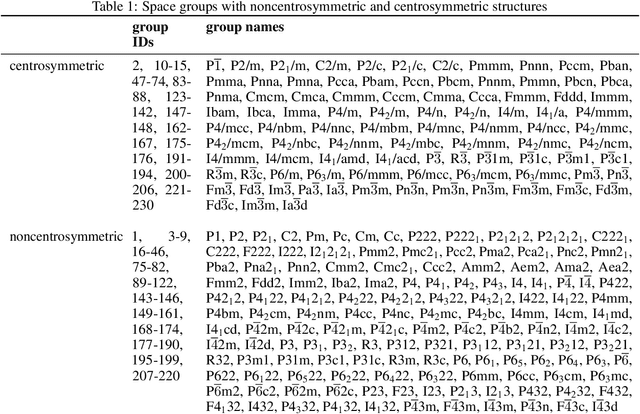
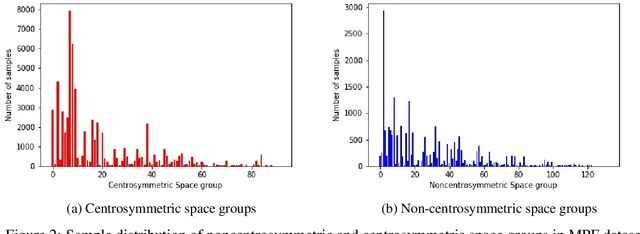
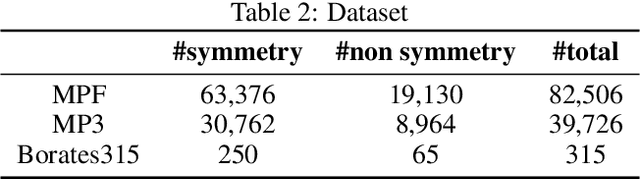
Noncentrosymmetric materials play a critical role in many important applications such as laser technology, communication systems,quantum computing, cybersecurity, and etc. However, the experimental discovery of new noncentrosymmetric materials is extremely difficult. Here we present a machine learning model that could predict whether the composition of a potential crystalline structure would be centrosymmetric or not. By evaluating a diverse set of composition features calculated using matminer featurizer package coupled with different machine learning algorithms, we find that Random Forest Classifiers give the best performance for noncentrosymmetric material prediction, reaching an accuracy of 84.8% when evaluated with 10 fold cross-validation on the dataset with 82,506 samples extracted from Materials Project. A random forest model trained with materials with only 3 elements gives even higher accuracy of 86.9%. We apply our ML model to screen potential noncentrosymmetric materials from 2,000,000 hypothetical materials generated by our inverse design engine and report the top 20 candidate noncentrosymmetric materials with 2 to 4 elements and top 20 borate candidates