 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAction-depedent Control Variates for Policy Optimization via Stein's Identity
Feb 23, 2018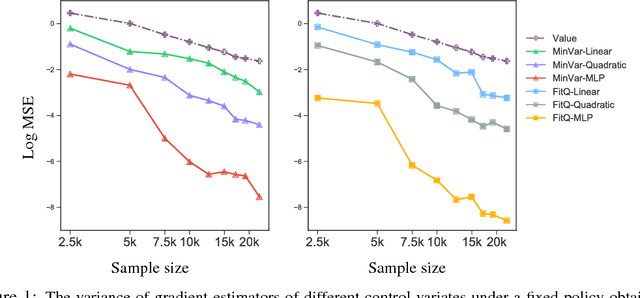

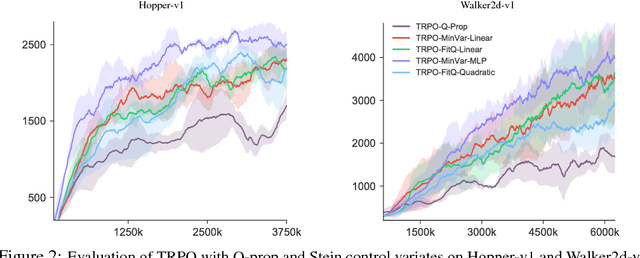
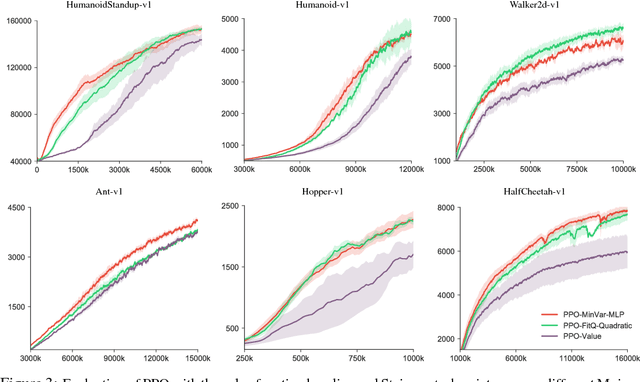
Policy gradient methods have achieved remarkable successes in solving challenging reinforcement learning problems. However, it still often suffers from the large variance issue on policy gradient estimation, which leads to poor sample efficiency during training. In this work, we propose a control variate method to effectively reduce variance for policy gradient methods. Motivated by the Stein's identity, our method extends the previous control variate methods used in REINFORCE and advantage actor-critic by introducing more general action-dependent baseline functions. Empirical studies show that our method significantly improves the sample efficiency of the state-of-the-art policy gradient approaches.
Empower Sequence Labeling with Task-Aware Neural Language Model
Nov 23, 2017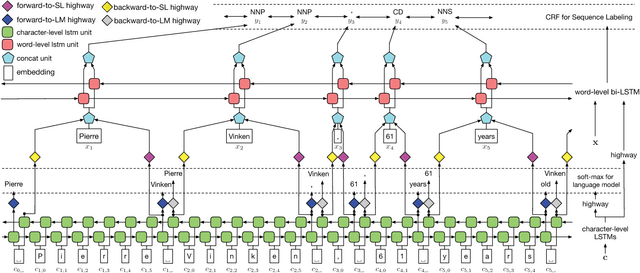

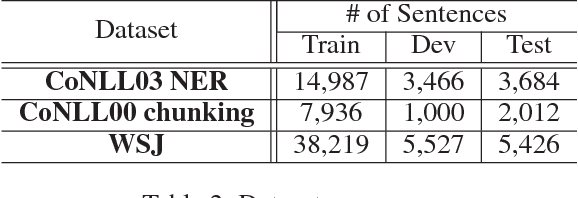
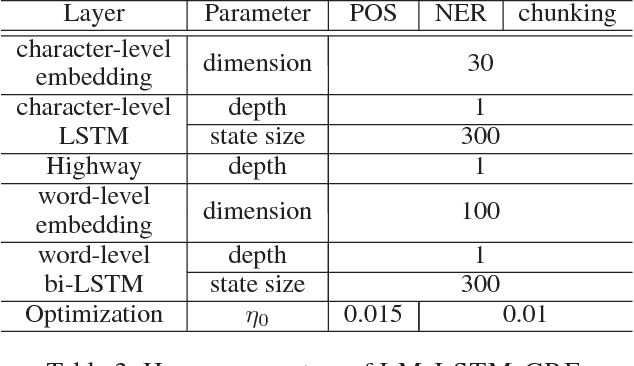
Linguistic sequence labeling is a general modeling approach that encompasses a variety of problems, such as part-of-speech tagging and named entity recognition. Recent advances in neural networks (NNs) make it possible to build reliable models without handcrafted features. However, in many cases, it is hard to obtain sufficient annotations to train these models. In this study, we develop a novel neural framework to extract abundant knowledge hidden in raw texts to empower the sequence labeling task. Besides word-level knowledge contained in pre-trained word embeddings, character-aware neural language models are incorporated to extract character-level knowledge. Transfer learning techniques are further adopted to mediate different components and guide the language model towards the key knowledge. Comparing to previous methods, these task-specific knowledge allows us to adopt a more concise model and conduct more efficient training. Different from most transfer learning methods, the proposed framework does not rely on any additional supervision. It extracts knowledge from self-contained order information of training sequences. Extensive experiments on benchmark datasets demonstrate the effectiveness of leveraging character-level knowledge and the efficiency of co-training. For example, on the CoNLL03 NER task, model training completes in about 6 hours on a single GPU, reaching F1 score of 91.71$\pm$0.10 without using any extra annotation.
Efficient Localized Inference for Large Graphical Models
Oct 28, 2017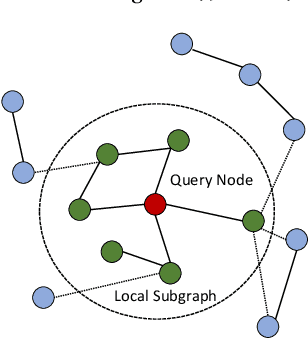
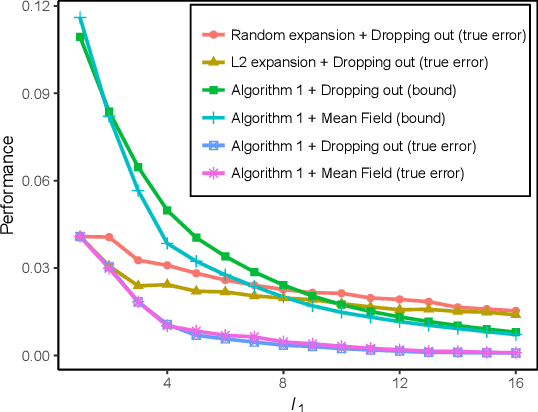
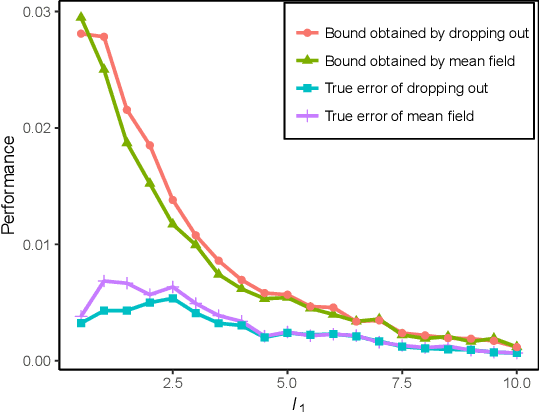
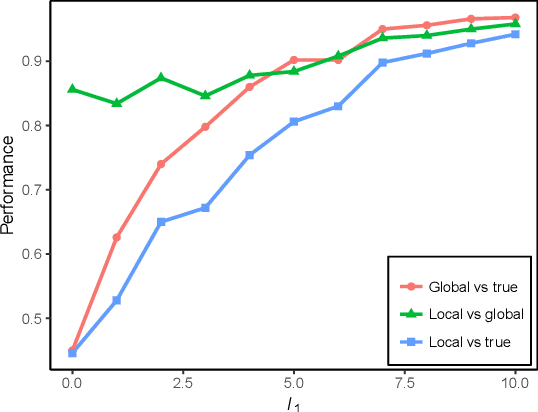
We propose a new localized inference algorithm for answering marginalization queries in large graphical models with the correlation decay property. Given a query variable and a large graphical model, we define a much smaller model in a local region around the query variable in the target model so that the marginal distribution of the query variable can be accurately approximated. We introduce two approximation error bounds based on the Dobrushin's comparison theorem and apply our bounds to derive a greedy expansion algorithm that efficiently guides the selection of neighbor nodes for localized inference. We verify our theoretical bounds on various datasets and demonstrate that our localized inference algorithm can provide fast and accurate approximation for large graphical models.
Energy-efficient Amortized Inference with Cascaded Deep Classifiers
Oct 10, 2017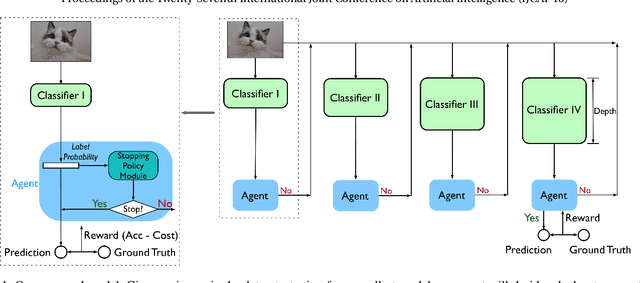
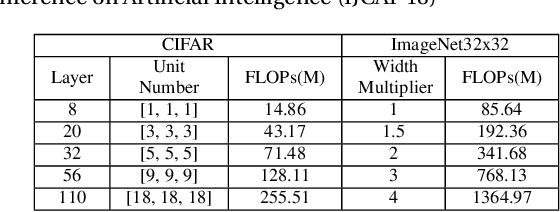

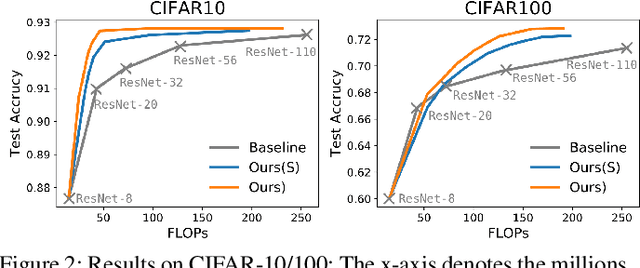
Deep neural networks have been remarkable successful in various AI tasks but often cast high computation and energy cost for energy-constrained applications such as mobile sensing. We address this problem by proposing a novel framework that optimizes the prediction accuracy and energy cost simultaneously, thus enabling effective cost-accuracy trade-off at test time. In our framework, each data instance is pushed into a cascade of deep neural networks with increasing sizes, and a selection module is used to sequentially determine when a sufficiently accurate classifier can be used for this data instance. The cascade of neural networks and the selection module are jointly trained in an end-to-end fashion by the REINFORCE algorithm to optimize a trade-off between the computational cost and the predictive accuracy. Our method is able to simultaneously improve the accuracy and efficiency by learning to assign easy instances to fast yet sufficiently accurate classifiers to save computation and energy cost, while assigning harder instances to deeper and more powerful classifiers to ensure satisfiable accuracy. With extensive experiments on several image classification datasets using cascaded ResNet classifiers, we demonstrate that our method outperforms the standard well-trained ResNets in accuracy but only requires less than 20% and 50% FLOPs cost on the CIFAR-10/100 datasets and 66% on the ImageNet dataset, respectively.
On the Selective and Invariant Representation of DCNN for High-Resolution Remote Sensing Image Recognition
Aug 04, 2017
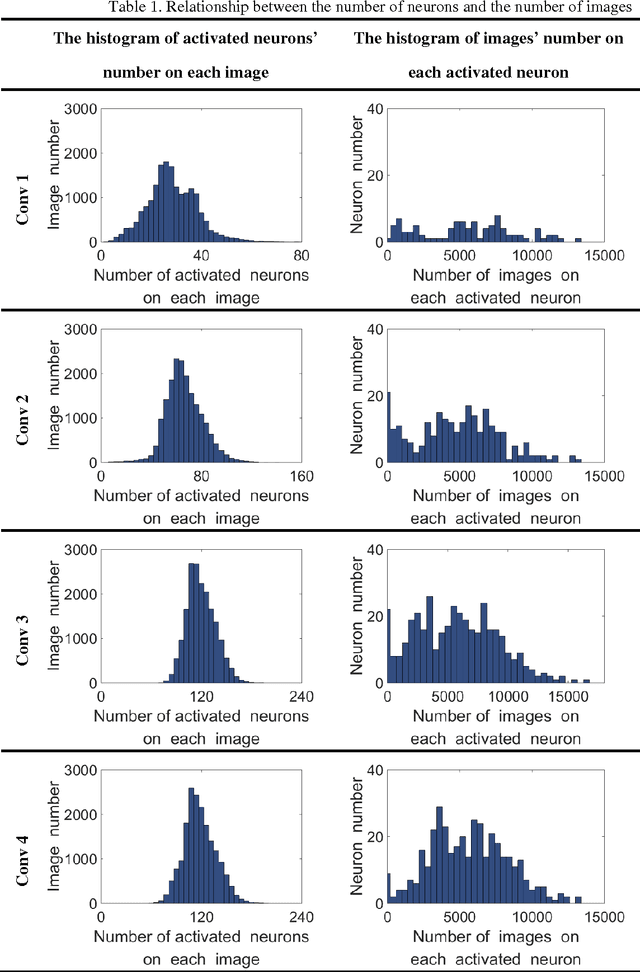
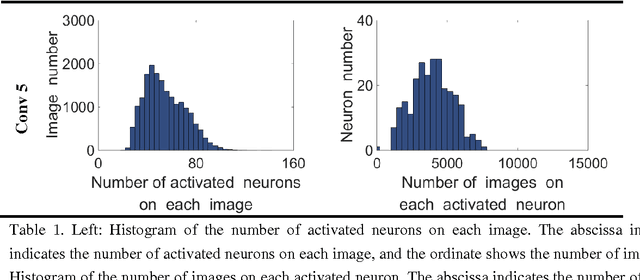
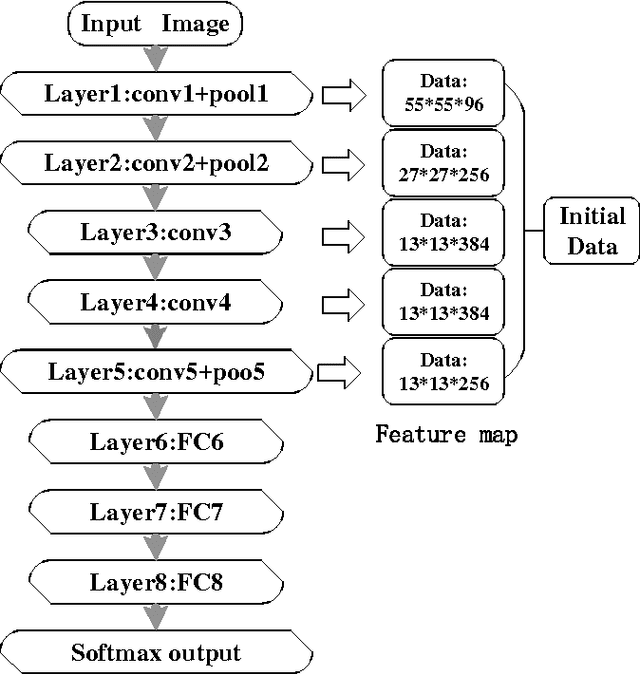
Human vision possesses strong invariance in image recognition. The cognitive capability of deep convolutional neural network (DCNN) is close to the human visual level because of hierarchical coding directly from raw image. Owing to its superiority in feature representation, DCNN has exhibited remarkable performance in scene recognition of high-resolution remote sensing (HRRS) images and classification of hyper-spectral remote sensing images. In-depth investigation is still essential for understanding why DCNN can accurately identify diverse ground objects via its effective feature representation. Thus, we train the deep neural network called AlexNet on our large scale remote sensing image recognition benchmark. At the neuron level in each convolution layer, we analyze the general properties of DCNN in HRRS image recognition by use of a framework of visual stimulation-characteristic response combined with feature coding-classification decoding. Specifically, we use histogram statistics, representational dissimilarity matrix, and class activation mapping to observe the selective and invariance representations of DCNN in HRRS image recognition. We argue that selective and invariance representations play important roles in remote sensing images tasks, such as classification, detection, and segment. Also selective and invariance representations are significant to design new DCNN liked models for analyzing and understanding remote sensing images.
What do We Learn by Semantic Scene Understanding for Remote Sensing imagery in CNN framework?
May 19, 2017
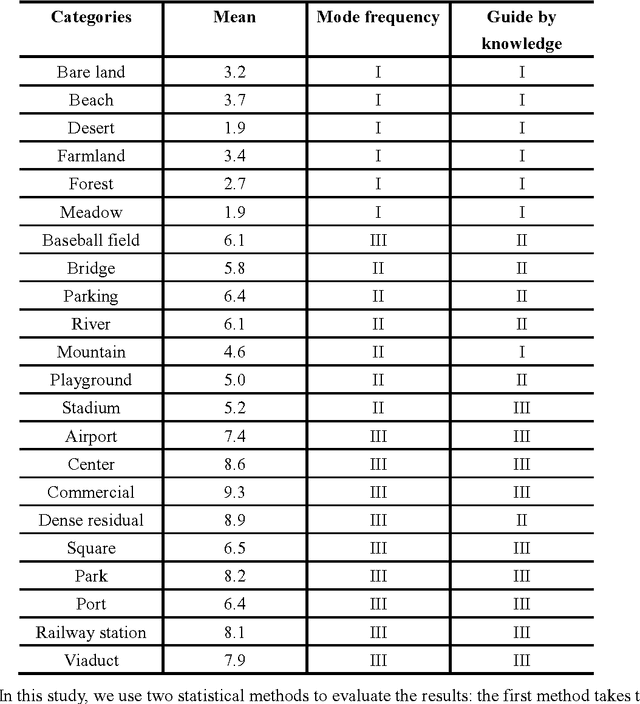
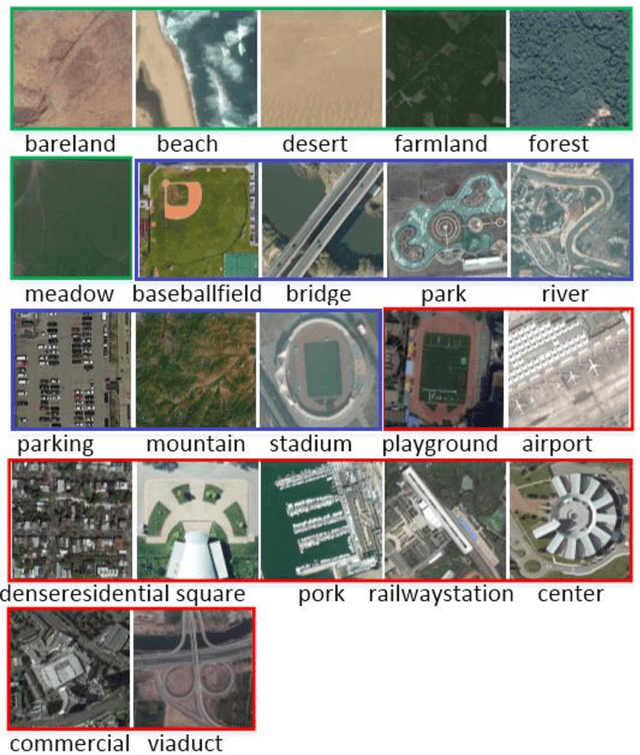

Recently, deep convolutional neural network (DCNN) achieved increasingly remarkable success and rapidly developed in the field of natural image recognition. Compared with the natural image, the scale of remote sensing image is larger and the scene and the object it represents are more macroscopic. This study inquires whether remote sensing scene and natural scene recognitions differ and raises the following questions: What are the key factors in remote sensing scene recognition? Is the DCNN recognition mechanism centered on object recognition still applicable to the scenarios of remote sensing scene understanding? We performed several experiments to explore the influence of the DCNN structure and the scale of remote sensing scene understanding from the perspective of scene complexity. Our experiment shows that understanding a complex scene depends on an in-depth network and multiple-scale perception. Using a visualization method, we qualitatively and quantitatively analyze the recognition mechanism in a complex remote sensing scene and demonstrate the importance of multi-objective joint semantic support.
Stein Variational Policy Gradient
Apr 07, 2017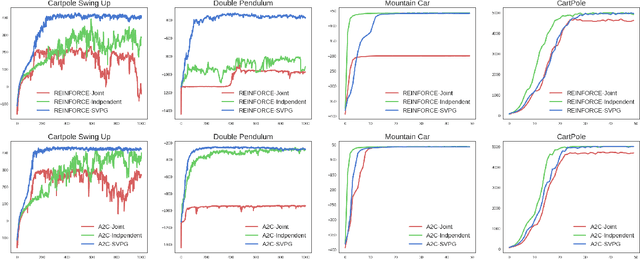
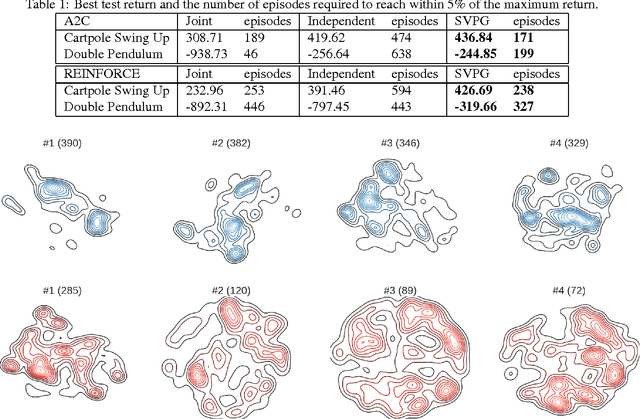
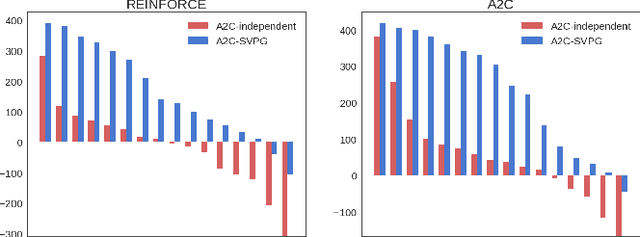
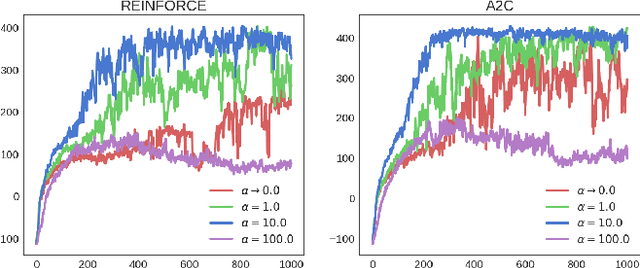
Policy gradient methods have been successfully applied to many complex reinforcement learning problems. However, policy gradient methods suffer from high variance, slow convergence, and inefficient exploration. In this work, we introduce a maximum entropy policy optimization framework which explicitly encourages parameter exploration, and show that this framework can be reduced to a Bayesian inference problem. We then propose a novel Stein variational policy gradient method (SVPG) which combines existing policy gradient methods and a repulsive functional to generate a set of diverse but well-behaved policies. SVPG is robust to initialization and can easily be implemented in a parallel manner. On continuous control problems, we find that implementing SVPG on top of REINFORCE and advantage actor-critic algorithms improves both average return and data efficiency.
On the Interpretability of Conditional Probability Estimates in the Agnostic Setting
Feb 28, 2017

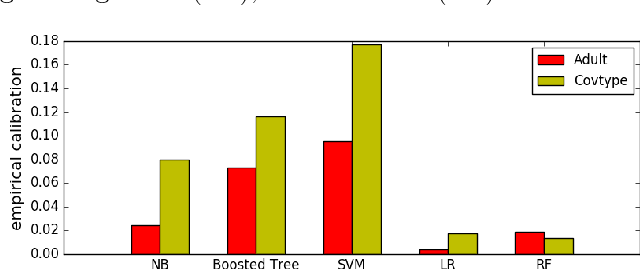
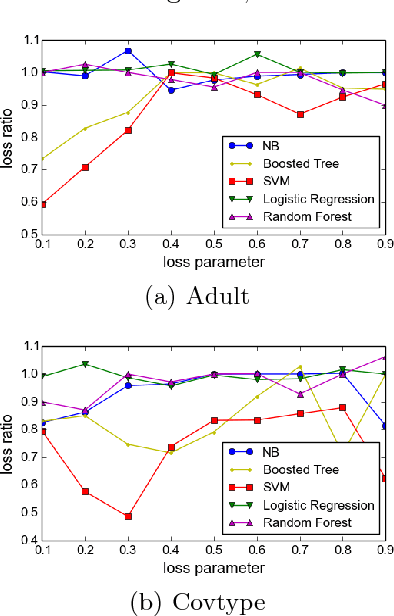
We study the interpretability of conditional probability estimates for binary classification under the agnostic setting or scenario. Under the agnostic setting, conditional probability estimates do not necessarily reflect the true conditional probabilities. Instead, they have a certain calibration property: among all data points that the classifier has predicted P(Y = 1|X) = p, p portion of them actually have label Y = 1. For cost-sensitive decision problems, this calibration property provides adequate support for us to use Bayes Decision Theory. In this paper, we define a novel measure for the calibration property together with its empirical counterpart, and prove an uniform convergence result between them. This new measure enables us to formally justify the calibration property of conditional probability estimations, and provides new insights on the problem of estimating and calibrating conditional probabilities.
Learning to Play in a Day: Faster Deep Reinforcement Learning by Optimality Tightening
Nov 05, 2016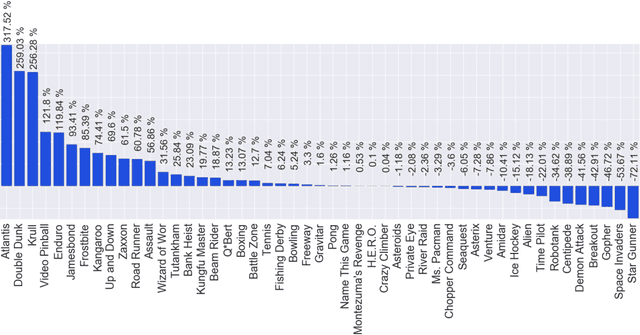

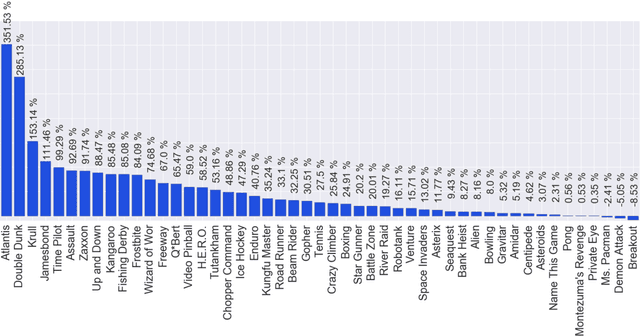
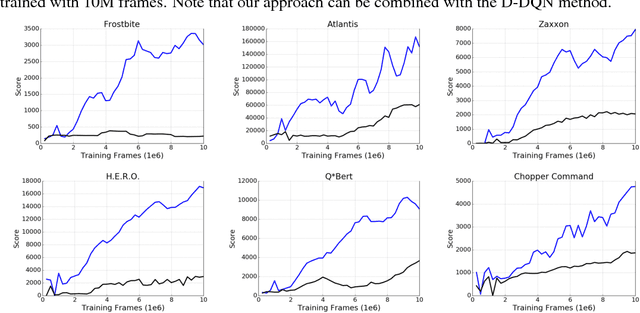
We propose a novel training algorithm for reinforcement learning which combines the strength of deep Q-learning with a constrained optimization approach to tighten optimality and encourage faster reward propagation. Our novel technique makes deep reinforcement learning more practical by drastically reducing the training time. We evaluate the performance of our approach on the 49 games of the challenging Arcade Learning Environment, and report significant improvements in both training time and accuracy.
DPPred: An Effective Prediction Framework with Concise Discriminative Patterns
Oct 31, 2016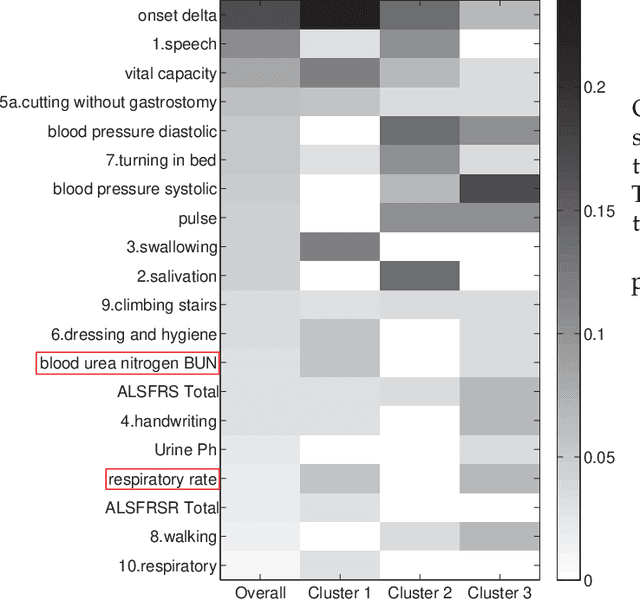
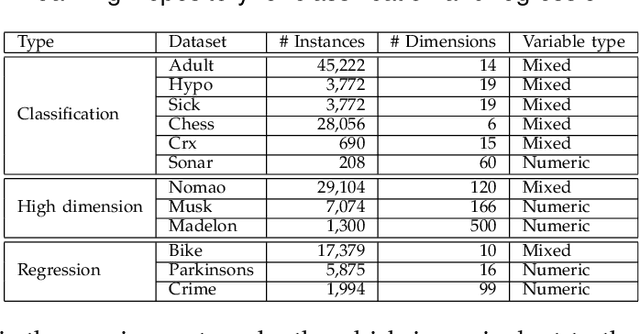
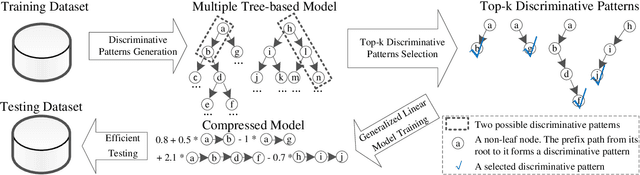

In the literature, two series of models have been proposed to address prediction problems including classification and regression. Simple models, such as generalized linear models, have ordinary performance but strong interpretability on a set of simple features. The other series, including tree-based models, organize numerical, categorical and high dimensional features into a comprehensive structure with rich interpretable information in the data. In this paper, we propose a novel Discriminative Pattern-based Prediction framework (DPPred) to accomplish the prediction tasks by taking their advantages of both effectiveness and interpretability. Specifically, DPPred adopts the concise discriminative patterns that are on the prefix paths from the root to leaf nodes in the tree-based models. DPPred selects a limited number of the useful discriminative patterns by searching for the most effective pattern combination to fit generalized linear models. Extensive experiments show that in many scenarios, DPPred provides competitive accuracy with the state-of-the-art as well as the valuable interpretability for developers and experts. In particular, taking a clinical application dataset as a case study, our DPPred outperforms the baselines by using only 40 concise discriminative patterns out of a potentially exponentially large set of patterns.