 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHierarchical Graph Representations in Digital Pathology
Mar 17, 2021
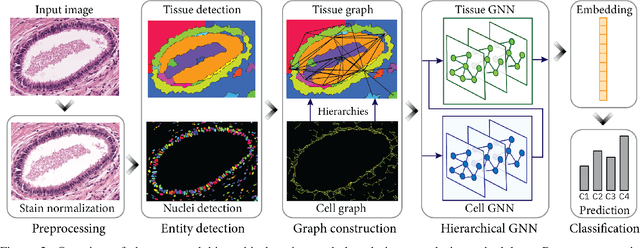
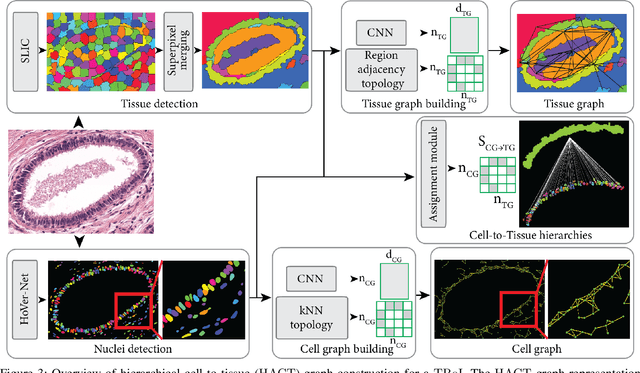
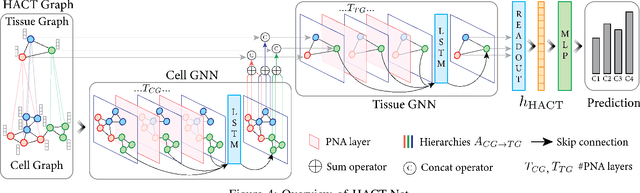
Cancer diagnosis, prognosis, and therapy response predictions from tissue specimens highly depend on the phenotype and topological distribution of constituting histological entities. Thus, adequate tissue representations for encoding histological entities is imperative for computer aided cancer patient care. To this end, several approaches have leveraged cell-graphs that encode cell morphology and organization to denote the tissue information. These allow for utilizing machine learning to map tissue representations to tissue functionality to help quantify their relationship. Though cellular information is crucial, it is incomplete alone to comprehensively characterize complex tissue structure. We herein treat the tissue as a hierarchical composition of multiple types of histological entities from fine to coarse level, capturing multivariate tissue information at multiple levels. We propose a novel multi-level hierarchical entity-graph representation of tissue specimens to model hierarchical compositions that encode histological entities as well as their intra- and inter-entity level interactions. Subsequently, a graph neural network is proposed to operate on the hierarchical entity-graph representation to map the tissue structure to tissue functionality. Specifically, for input histology images we utilize well-defined cells and tissue regions to build HierArchical Cell-to-Tissue (HACT) graph representations, and devise HACT-Net, a graph neural network, to classify such HACT representations. As part of this work, we introduce the BReAst Carcinoma Subtyping (BRACS) dataset, a large cohort of H&E stained breast tumor images, to evaluate our proposed methodology against pathologists and state-of-the-art approaches. Through comparative assessment and ablation studies, our method is demonstrated to yield superior classification results compared to alternative methods as well as pathologists.
Self-Attentive Spatial Adaptive Normalization for Cross-Modality Domain Adaptation
Mar 05, 2021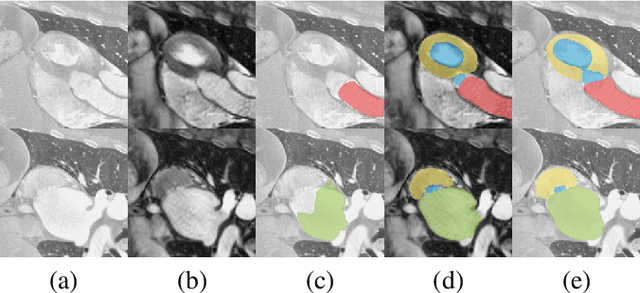
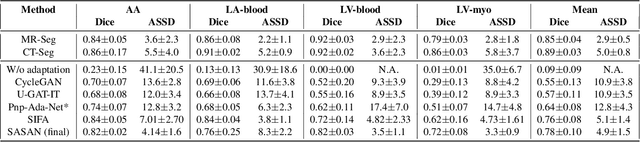
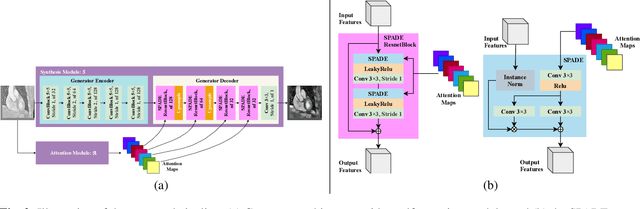
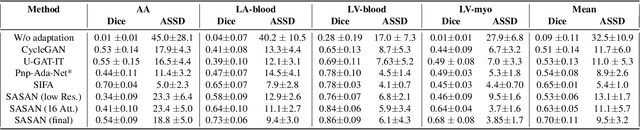
Despite the successes of deep neural networks on many challenging vision tasks, they often fail to generalize to new test domains that are not distributed identically to the training data. The domain adaptation becomes more challenging for cross-modality medical data with a notable domain shift. Given that specific annotated imaging modalities may not be accessible nor complete. Our proposed solution is based on the cross-modality synthesis of medical images to reduce the costly annotation burden by radiologists and bridge the domain gap in radiological images. We present a novel approach for image-to-image translation in medical images, capable of supervised or unsupervised (unpaired image data) setups. Built upon adversarial training, we propose a learnable self-attentive spatial normalization of the deep convolutional generator network's intermediate activations. Unlike previous attention-based image-to-image translation approaches, which are either domain-specific or require distortion of the source domain's structures, we unearth the importance of the auxiliary semantic information to handle the geometric changes and preserve anatomical structures during image translation. We achieve superior results for cross-modality segmentation between unpaired MRI and CT data for multi-modality whole heart and multi-modal brain tumor MRI (T1/T2) datasets compared to the state-of-the-art methods. We also observe encouraging results in cross-modality conversion for paired MRI and CT images on a brain dataset. Furthermore, a detailed analysis of the cross-modality image translation, thorough ablation studies confirm our proposed method's efficacy.
Learning Whole-Slide Segmentation from Inexact and Incomplete Labels using Tissue Graphs
Mar 04, 2021Segmenting histology images into diagnostically relevant regions is imperative to support timely and reliable decisions by pathologists. To this end, computer-aided techniques have been proposed to delineate relevant regions in scanned histology slides. However, the techniques necessitate task-specific large datasets of annotated pixels, which is tedious, time-consuming, expensive, and infeasible to acquire for many histology tasks. Thus, weakly-supervised semantic segmentation techniques are proposed to utilize weak supervision that is cheaper and quicker to acquire. In this paper, we propose SegGini, a weakly supervised segmentation method using graphs, that can utilize weak multiplex annotations, i.e. inexact and incomplete annotations, to segment arbitrary and large images, scaling from tissue microarray (TMA) to whole slide image (WSI). Formally, SegGini constructs a tissue-graph representation for an input histology image, where the graph nodes depict tissue regions. Then, it performs weakly-supervised segmentation via node classification by using inexact image-level labels, incomplete scribbles, or both. We evaluated SegGini on two public prostate cancer datasets containing TMAs and WSIs. Our method achieved state-of-the-art segmentation performance on both datasets for various annotation settings while being comparable to a pathologist baseline.
Self-Taught Semi-Supervised Anomaly Detection on Upper Limb X-rays
Feb 22, 2021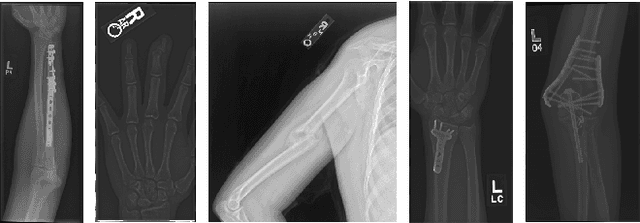
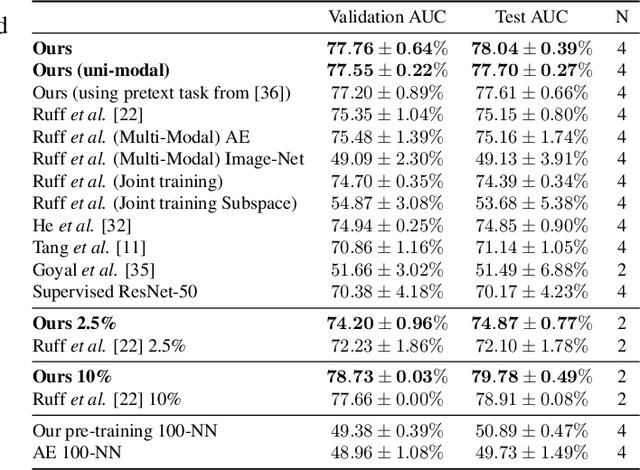
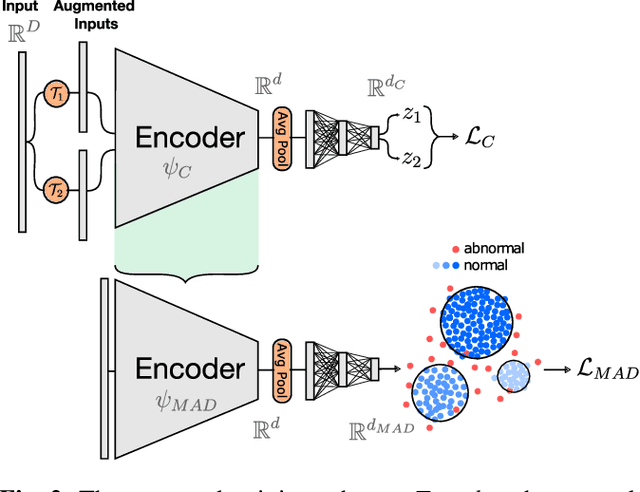
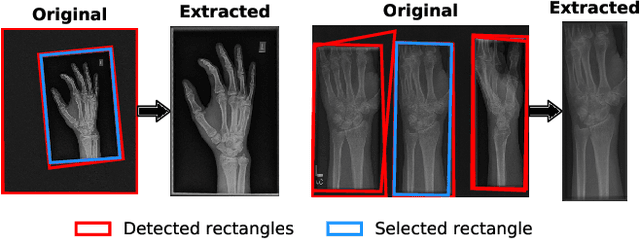
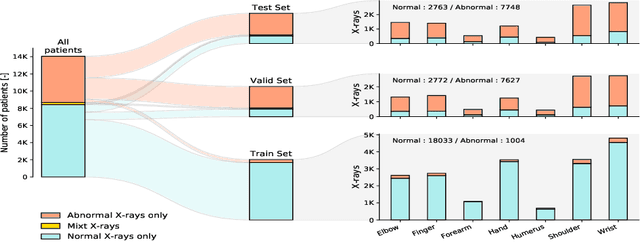
Detecting anomalies in musculoskeletal radiographs is of paramount importance for large-scale screening in the radiology workflow. Supervised deep networks take for granted a large number of annotations by radiologists, which is often prohibitively very time-consuming to acquire. Moreover, supervised systems are tailored to closed set scenarios, e.g., trained models suffer from overfitting to previously seen rare anomalies at training. Instead, our approach's rationale is to use task agnostic pretext tasks to leverage unlabeled data based on a cross-sample similarity measure. Besides, we formulate a complex distribution of data from normal class within our framework to avoid a potential bias on the side of anomalies. Through extensive experiments, we show that our method outperforms baselines across unsupervised and self-supervised anomaly detection settings on a real-world medical dataset, the MURA dataset. We also provide rich ablation studies to analyze each training stage's effect and loss terms on the final performance.
Quantifying Explainers of Graph Neural Networks in Computational Pathology
Nov 25, 2020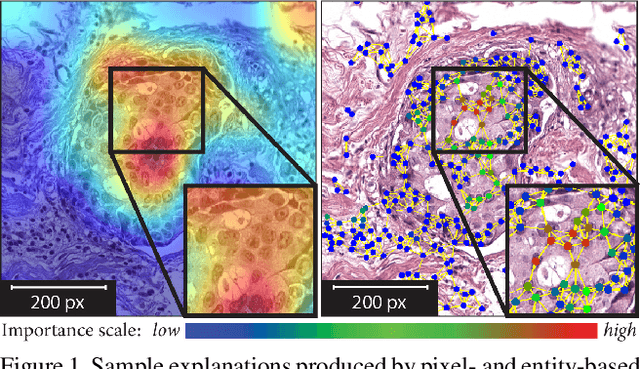
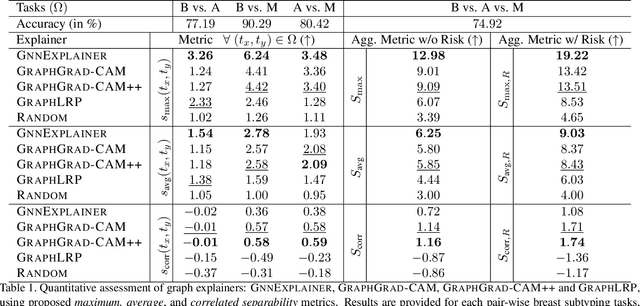
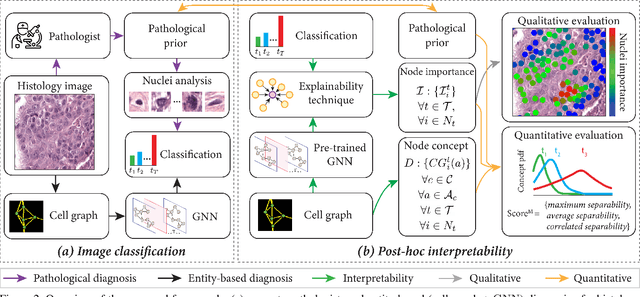
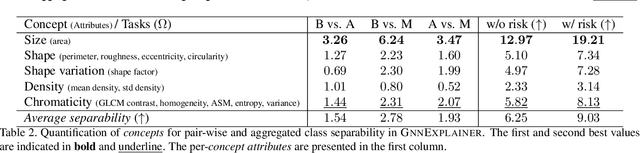
Explainability of deep learning methods is imperative to facilitate their clinical adoption in digital pathology. However, popular deep learning methods and explainability techniques (explainers) based on pixel-wise processing disregard biological entities' notion, thus complicating comprehension by pathologists. In this work, we address this by adopting biological entity-based graph processing and graph explainers enabling explanations accessible to pathologists. In this context, a major challenge becomes to discern meaningful explainers, particularly in a standardized and quantifiable fashion. To this end, we propose herein a set of novel quantitative metrics based on statistics of class separability using pathologically measurable concepts to characterize graph explainers. We employ the proposed metrics to evaluate three types of graph explainers, namely the layer-wise relevance propagation, gradient-based saliency, and graph pruning approaches, to explain Cell-Graph representations for Breast Cancer Subtyping. The proposed metrics are also applicable in other domains by using domain-specific intuitive concepts. We validate the qualitative and quantitative findings on the BRACS dataset, a large cohort of breast cancer RoIs, by expert pathologists.
Anomaly Detection on X-Rays Using Self-Supervised Aggregation Learning
Oct 19, 2020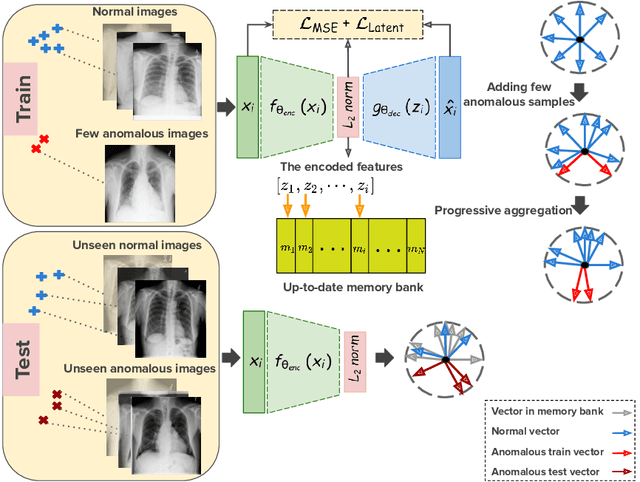
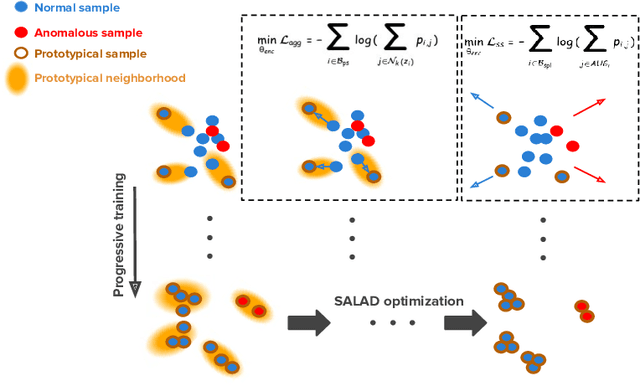
Deep anomaly detection models using a supervised mode of learning usually work under a closed set assumption and suffer from overfitting to previously seen rare anomalies at training, which hinders their applicability in a real scenario. In addition, obtaining annotations for X-rays is very time consuming and requires extensive training of radiologists. Hence, training anomaly detection in a fully unsupervised or self-supervised fashion would be advantageous, allowing a significant reduction of time spent on the report by radiologists. In this paper, we present SALAD, an end-to-end deep self-supervised methodology for anomaly detection on X-Ray images. The proposed method is based on an optimization strategy in which a deep neural network is encouraged to represent prototypical local patterns of the normal data in the embedding space. During training, we record the prototypical patterns of normal training samples via a memory bank. Our anomaly score is then derived by measuring similarity to a weighted combination of normal prototypical patterns within a memory bank without using any anomalous patterns. We present extensive experiments on the challenging NIH Chest X-rays and MURA dataset, which indicate that our algorithm improves state-of-the-art methods by a wide margin.
CNN-Based Ultrasound Image Reconstruction for Ultrafast Displacement Tracking
Sep 03, 2020
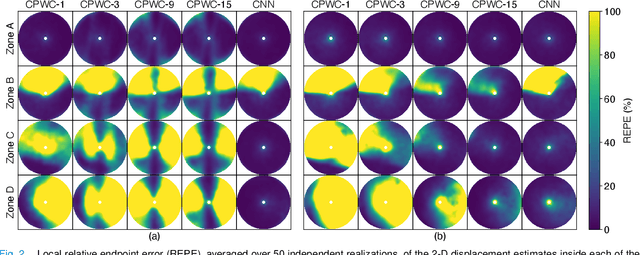
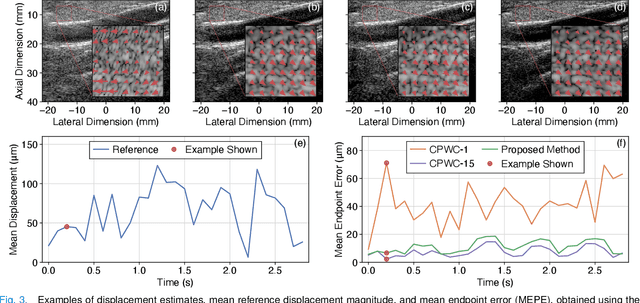
Thanks to its capability of acquiring full-view frames at multiple kilohertz, ultrafast ultrasound imaging unlocked the analysis of rapidly changing physical phenomena in the human body, with pioneering applications such as ultrasensitive flow imaging in the cardiovascular system or shear-wave elastography. The accuracy achievable with these motion estimation techniques is strongly contingent upon two contradictory requirements: a high quality of consecutive frames and a high frame rate. Indeed, the image quality can usually be improved by increasing the number of steered ultrafast acquisitions, but at the expense of a reduced frame rate and possible motion artifacts. To achieve accurate motion estimation at uncompromised frame rates and immune to motion artifacts, the proposed approach relies on single ultrafast acquisitions to reconstruct high-quality frames and on only two consecutive frames to obtain 2-D displacement estimates. To this end, we deployed a convolutional neural network-based image reconstruction method combined with a speckle tracking algorithm based on cross-correlation. Numerical and in vivo experiments, conducted in the context of plane-wave imaging, demonstrate that the proposed approach is capable of estimating displacements in regions where the presence of side lobe and grating lobe artifacts prevents any displacement estimation with a state-of-the-art technique that rely on conventional delay-and-sum beamforming. The proposed approach may therefore unlock the full potential of ultrafast ultrasound, in applications such as ultrasensitive cardiovascular motion and flow analysis or shear-wave elastography.
CNN-Based Image Reconstruction Method for Ultrafast Ultrasound Imaging
Aug 28, 2020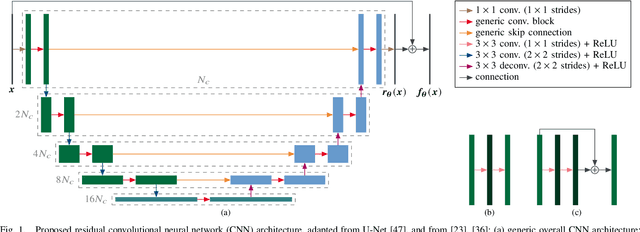
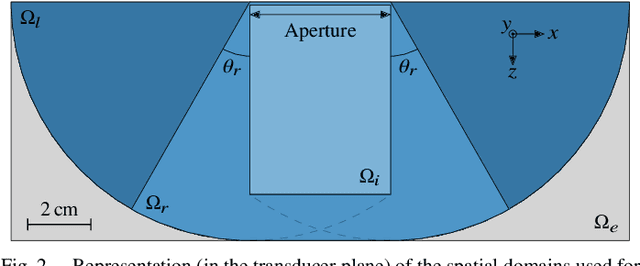
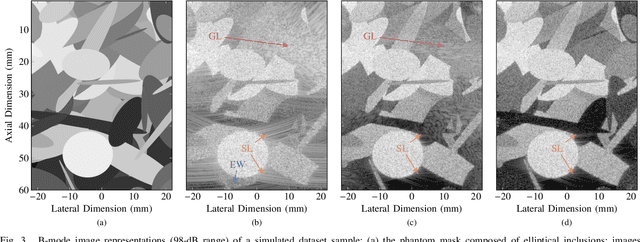
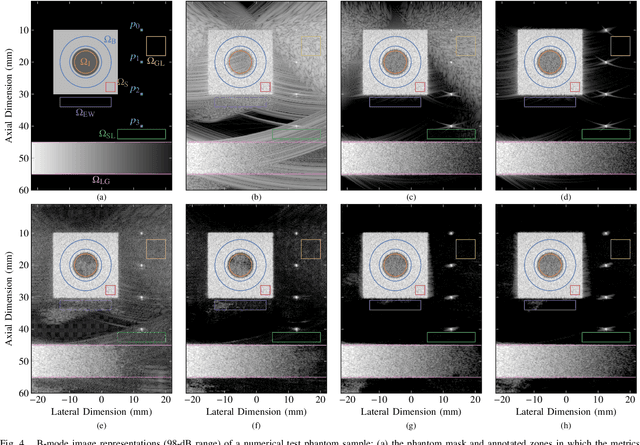
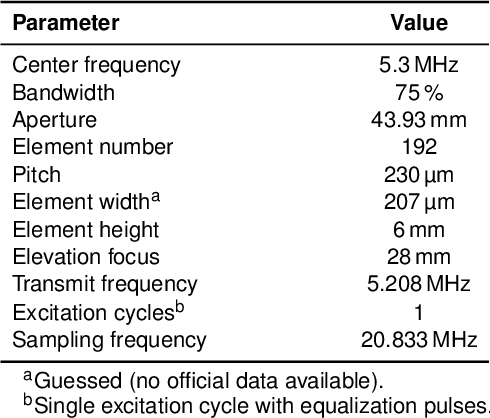
Ultrafast ultrasound (US) revolutionized biomedical imaging with its capability of acquiring full-view frames at over 1 kHz, unlocking breakthrough modalities such as shear-wave elastography and functional US neuroimaging. Yet, it suffers from strong diffraction artifacts, mainly caused by grating lobes, side lobes, or edge waves. Multiple acquisitions are typically required to obtain a sufficient image quality, at the cost of a reduced frame rate. To answer the increasing demand for high-quality imaging from single-shot acquisitions, we propose a two-step convolutional neural network (CNN)-based image reconstruction method, compatible with real-time imaging. A low-quality estimate is obtained by means of a backprojection-based operation, akin to conventional delay-and-sum beamforming, from which a high-quality image is restored using a residual CNN with multi-scale and multi-channel filtering properties, trained specifically to remove the diffraction artifacts inherent to ultrafast US imaging. To account for both the high dynamic range and the radio frequency property of US images, we introduce the mean signed logarithmic absolute error (MSLAE) as training loss function. Experiments were conducted with a linear transducer array, in single plane wave (PW) imaging. Trainings were performed on a simulated dataset, crafted to contain a wide diversity of structures and echogenicities. Extensive numerical evaluations demonstrate that the proposed approach can reconstruct images from single PWs with a quality similar to that of gold-standard synthetic aperture imaging, on a dynamic range in excess of 60 dB. In vitro and in vivo experiments show that trainings performed on simulated data translate well to experimental settings.
Automated Detection of Cortical Lesions in Multiple Sclerosis Patients with 7T MRI
Aug 15, 2020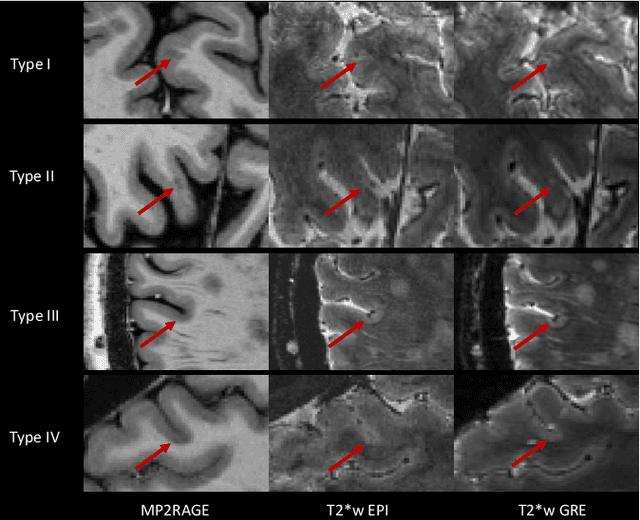
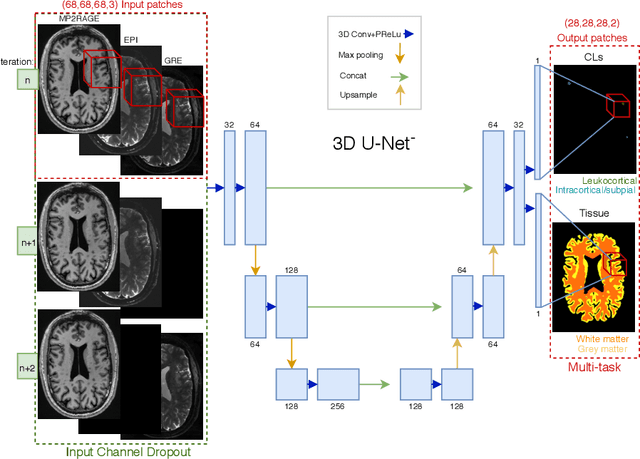
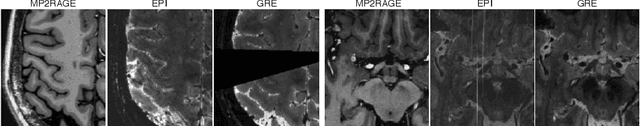
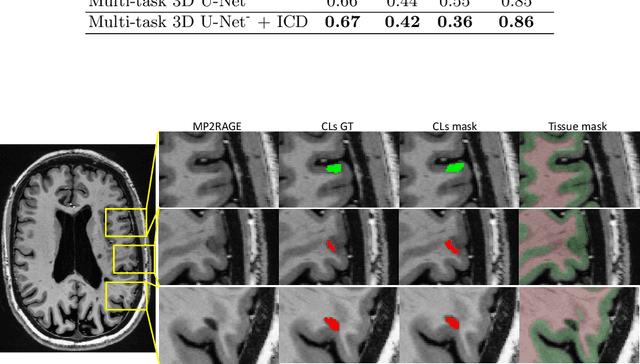
The automated detection of cortical lesions (CLs) in patients with multiple sclerosis (MS) is a challenging task that, despite its clinical relevance, has received very little attention. Accurate detection of the small and scarce lesions requires specialized sequences and high or ultra-high field MRI. For supervised training based on multimodal structural MRI at 7T, two experts generated ground truth segmentation masks of 60 patients with 2014 CLs. We implemented a simplified 3D U-Net with three resolution levels (3D U-Net-). By increasing the complexity of the task (adding brain tissue segmentation), while randomly dropping input channels during training, we improved the performance compared to the baseline. Considering a minimum lesion size of 0.75 {\mu}L, we achieved a lesion-wise cortical lesion detection rate of 67% and a false positive rate of 42%. However, 393 (24%) of the lesions reported as false positives were post-hoc confirmed as potential or definite lesions by an expert. This indicates the potential of the proposed method to support experts in the tedious process of CL manual segmentation.
Structure Preserving Stain Normalization of Histopathology Images Using Self-Supervised Semantic Guidance
Aug 05, 2020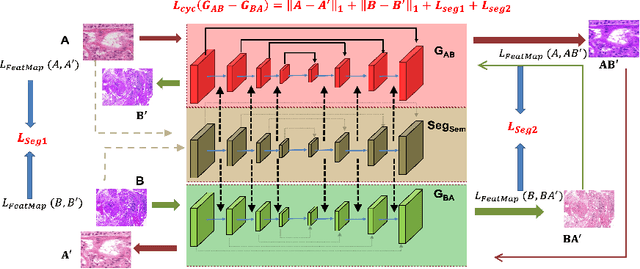
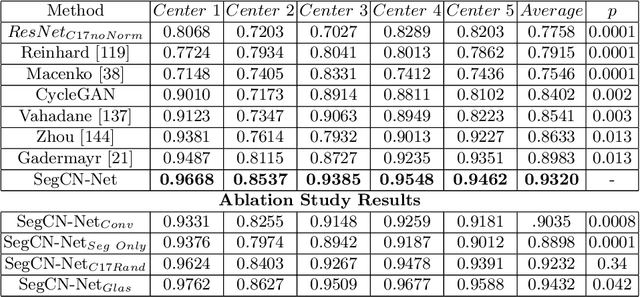
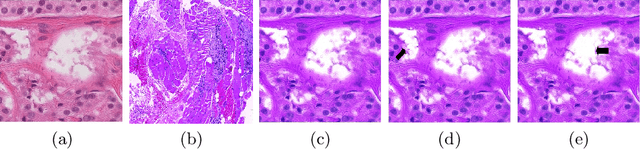
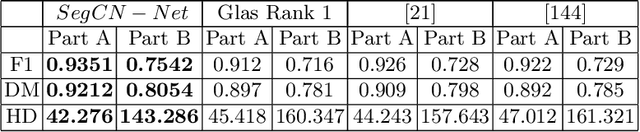
Although generative adversarial network (GAN) based style transfer is state of the art in histopathology color-stain normalization, they do not explicitly integrate structural information of tissues. We propose a self-supervised approach to incorporate semantic guidance into a GAN based stain normalization framework and preserve detailed structural information. Our method does not require manual segmentation maps which is a significant advantage over existing methods. We integrate semantic information at different layers between a pre-trained semantic network and the stain color normalization network. The proposed scheme outperforms other color normalization methods leading to better classification and segmentation performance.