 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAI Age Discrepancy: A Novel Parameter for Frailty Assessment in Kidney Tumor Patients
Jul 02, 2024Kidney cancer is a global health concern, and accurate assessment of patient frailty is crucial for optimizing surgical outcomes. This paper introduces AI Age Discrepancy, a novel metric derived from machine learning analysis of preoperative abdominal CT scans, as a potential indicator of frailty and postoperative risk in kidney cancer patients. This retrospective study of 599 patients from the 2023 Kidney Tumor Segmentation (KiTS) challenge dataset found that a higher AI Age Discrepancy is significantly associated with longer hospital stays and lower overall survival rates, independent of established factors. This suggests that AI Age Discrepancy may provide valuable insights into patient frailty and could thus inform clinical decision-making in kidney cancer treatment.
Efficient Parameter Optimisation for Quantum Kernel Alignment: A Sub-sampling Approach in Variational Training
Jan 05, 2024



Quantum machine learning with quantum kernels for classification problems is a growing area of research. Recently, quantum kernel alignment techniques that parameterise the kernel have been developed, allowing the kernel to be trained and therefore aligned with a specific dataset. While quantum kernel alignment is a promising technique, it has been hampered by considerable training costs because the full kernel matrix must be constructed at every training iteration. Addressing this challenge, we introduce a novel method that seeks to balance efficiency and performance. We present a sub-sampling training approach that uses a subset of the kernel matrix at each training step, thereby reducing the overall computational cost of the training. In this work, we apply the sub-sampling method to synthetic datasets and a real-world breast cancer dataset and demonstrate considerable reductions in the number of circuits required to train the quantum kernel while maintaining classification accuracy.
Weakly Supervised Joint Whole-Slide Segmentation and Classification in Prostate Cancer
Jan 07, 2023The segmentation and automatic identification of histological regions of diagnostic interest offer a valuable aid to pathologists. However, segmentation methods are hampered by the difficulty of obtaining pixel-level annotations, which are tedious and expensive to obtain for Whole-Slide images (WSI). To remedy this, weakly supervised methods have been developed to exploit the annotations directly available at the image level. However, to our knowledge, none of these techniques is adapted to deal with WSIs. In this paper, we propose WholeSIGHT, a weakly-supervised method, to simultaneously segment and classify WSIs of arbitrary shapes and sizes. Formally, WholeSIGHT first constructs a tissue-graph representation of the WSI, where the nodes and edges depict tissue regions and their interactions, respectively. During training, a graph classification head classifies the WSI and produces node-level pseudo labels via post-hoc feature attribution. These pseudo labels are then used to train a node classification head for WSI segmentation. During testing, both heads simultaneously render class prediction and segmentation for an input WSI. We evaluated WholeSIGHT on three public prostate cancer WSI datasets. Our method achieved state-of-the-art weakly-supervised segmentation performance on all datasets while resulting in better or comparable classification with respect to state-of-the-art weakly-supervised WSI classification methods. Additionally, we quantify the generalization capability of our method in terms of segmentation and classification performance, uncertainty estimation, and model calibration.
Differentiable Zooming for Multiple Instance Learning on Whole-Slide Images
Apr 30, 2022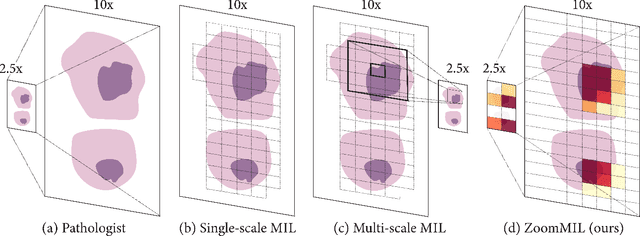
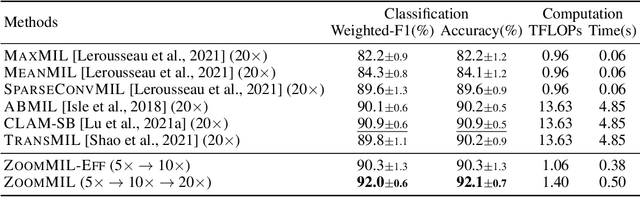
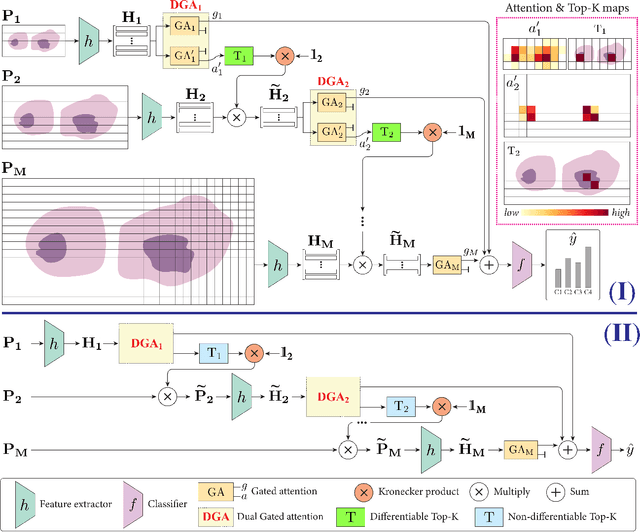
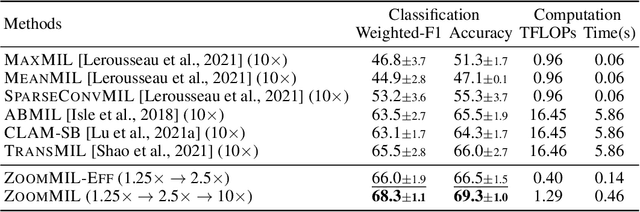
Multiple Instance Learning (MIL) methods have become increasingly popular for classifying giga-pixel sized Whole-Slide Images (WSIs) in digital pathology. Most MIL methods operate at a single WSI magnification, by processing all the tissue patches. Such a formulation induces high computational requirements, and constrains the contextualization of the WSI-level representation to a single scale. A few MIL methods extend to multiple scales, but are computationally more demanding. In this paper, inspired by the pathological diagnostic process, we propose ZoomMIL, a method that learns to perform multi-level zooming in an end-to-end manner. ZoomMIL builds WSI representations by aggregating tissue-context information from multiple magnifications. The proposed method outperforms the state-of-the-art MIL methods in WSI classification on two large datasets, while significantly reducing the computational demands with regard to Floating-Point Operations (FLOPs) and processing time by up to 40x.
BRACS: A Dataset for BReAst Carcinoma Subtyping in H&E Histology Images
Nov 08, 2021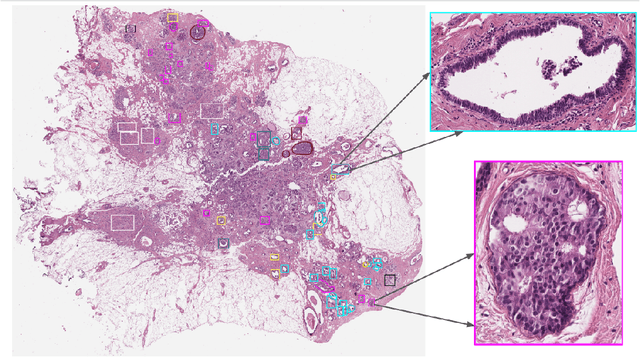

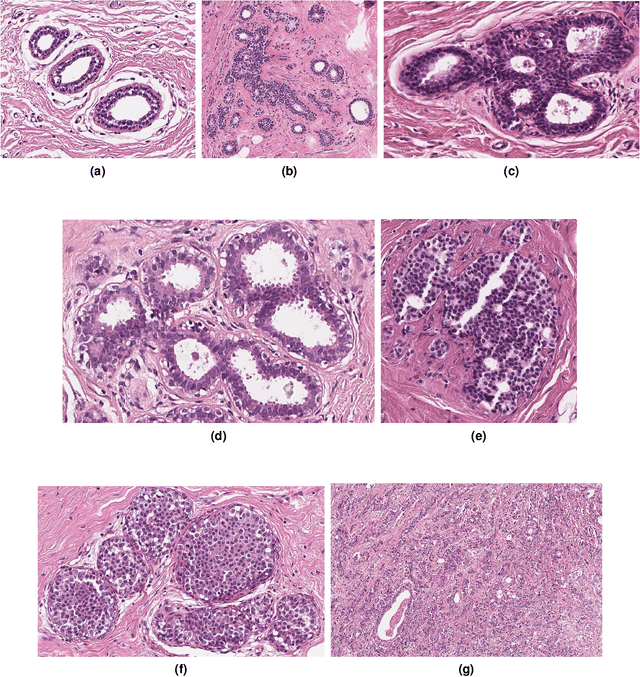
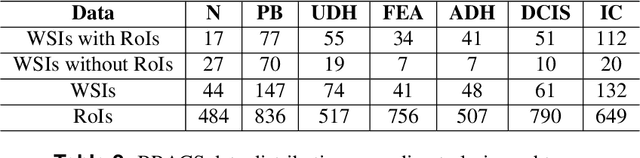
Breast cancer is the most commonly diagnosed cancer and registers the highest number of deaths for women with cancer. Recent advancements in diagnostic activities combined with large-scale screening policies have significantly lowered the mortality rates for breast cancer patients. However, the manual inspection of tissue slides by the pathologists is cumbersome, time-consuming, and is subject to significant inter- and intra-observer variability. Recently, the advent of whole-slide scanning systems have empowered the rapid digitization of pathology slides, and enabled to develop digital workflows. These advances further enable to leverage Artificial Intelligence (AI) to assist, automate, and augment pathological diagnosis. But the AI techniques, especially Deep Learning (DL), require a large amount of high-quality annotated data to learn from. Constructing such task-specific datasets poses several challenges, such as, data-acquisition level constrains, time-consuming and expensive annotations, and anonymization of private information. In this paper, we introduce the BReAst Carcinoma Subtyping (BRACS) dataset, a large cohort of annotated Hematoxylin & Eosin (H&E)-stained images to facilitate the characterization of breast lesions. BRACS contains 547 Whole-Slide Images (WSIs), and 4539 Regions of Interest (ROIs) extracted from the WSIs. Each WSI, and respective ROIs, are annotated by the consensus of three board-certified pathologists into different lesion categories. Specifically, BRACS includes three lesion types, i.e., benign, malignant and atypical, which are further subtyped into seven categories. It is, to the best of our knowledge, the largest annotated dataset for breast cancer subtyping both at WSI- and ROI-level. Further, by including the understudied atypical lesions, BRACS offers an unique opportunity for leveraging AI to better understand their characteristics.
HistoCartography: A Toolkit for Graph Analytics in Digital Pathology
Jul 21, 2021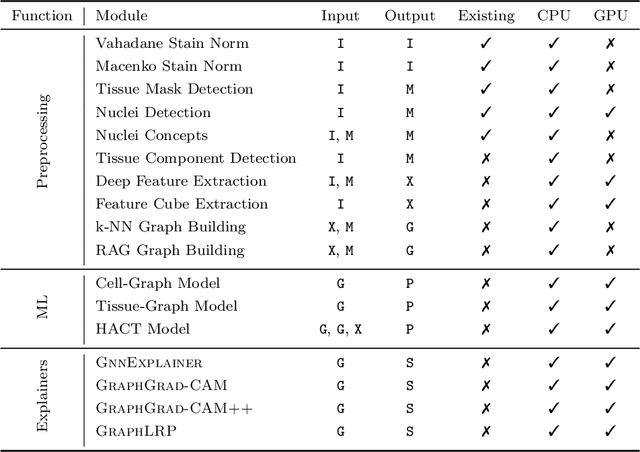
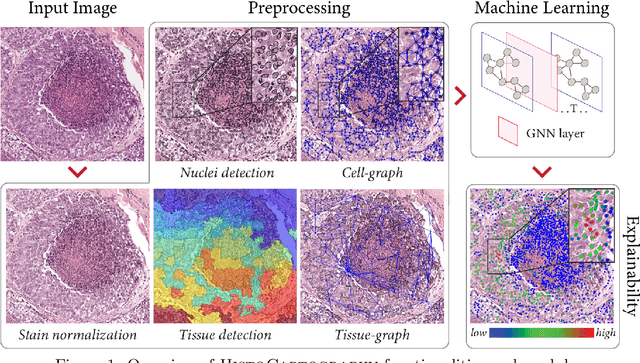
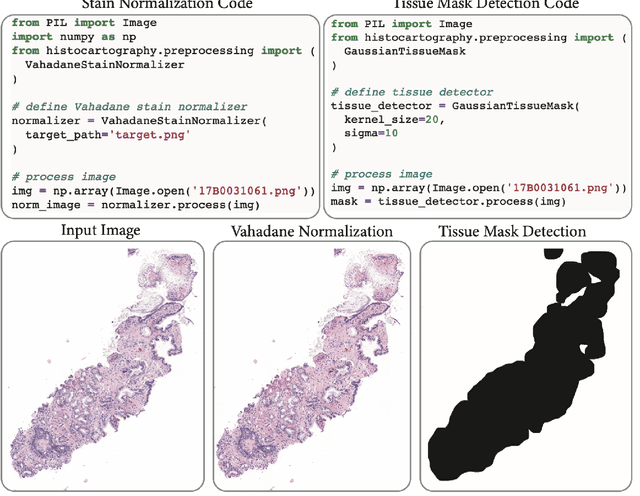
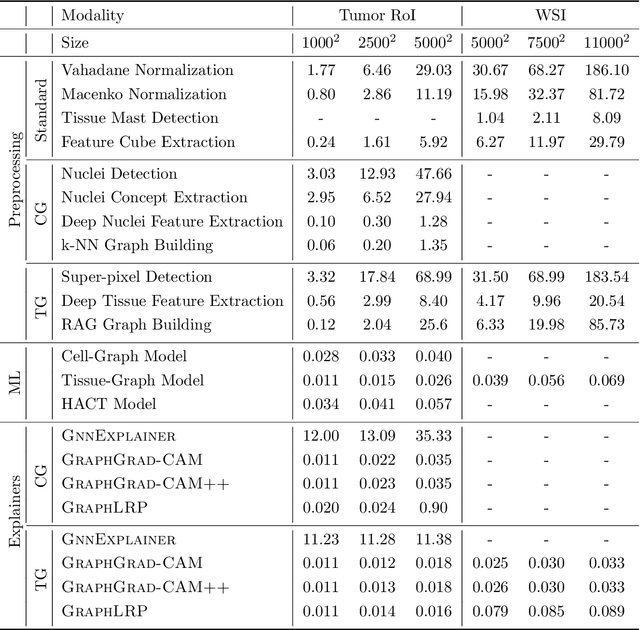
Advances in entity-graph based analysis of histopathology images have brought in a new paradigm to describe tissue composition, and learn the tissue structure-to-function relationship. Entity-graphs offer flexible and scalable representations to characterize tissue organization, while allowing the incorporation of prior pathological knowledge to further support model interpretability and explainability. However, entity-graph analysis requires prerequisites for image-to-graph translation and knowledge of state-of-the-art machine learning algorithms applied to graph-structured data, which can potentially hinder their adoption. In this work, we aim to alleviate these issues by developing HistoCartography, a standardized python API with necessary preprocessing, machine learning and explainability tools to facilitate graph-analytics in computational pathology. Further, we have benchmarked the computational time and performance on multiple datasets across different imaging types and histopathology tasks to highlight the applicability of the API for building computational pathology workflows.
Match What Matters: Generative Implicit Feature Replay for Continual Learning
Jun 09, 2021
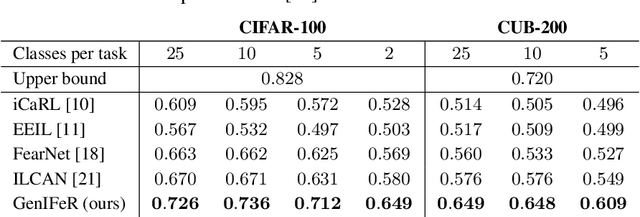

Neural networks are prone to catastrophic forgetting when trained incrementally on different tasks. In order to prevent forgetting, most existing methods retain a small subset of previously seen samples, which in turn can be used for joint training with new tasks. While this is indeed effective, it may not always be possible to store such samples, e.g., due to data protection regulations. In these cases, one can instead employ generative models to create artificial samples or features representing memories from previous tasks. Following a similar direction, we propose GenIFeR (Generative Implicit Feature Replay) for class-incremental learning. The main idea is to train a generative adversarial network (GAN) to generate images that contain realistic features. While the generator creates images at full resolution, the discriminator only sees the corresponding features extracted by the continually trained classifier. Since the classifier compresses raw images into features that are actually relevant for classification, the GAN can match this target distribution more accurately. On the other hand, allowing the generator to create full resolution images has several benefits: In contrast to previous approaches, the feature extractor of the classifier does not have to be frozen. In addition, we can employ augmentations on generated images, which not only boosts classification performance, but also mitigates discriminator overfitting during GAN training. We empirically show that GenIFeR is superior to both conventional generative image and feature replay. In particular, we significantly outperform the state-of-the-art in generative replay for various settings on the CIFAR-100 and CUB-200 datasets.
Hierarchical Graph Representations in Digital Pathology
Mar 17, 2021
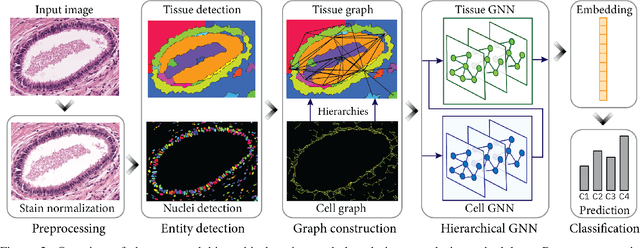
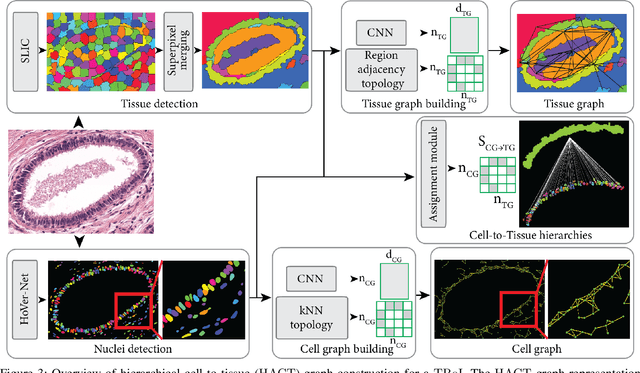
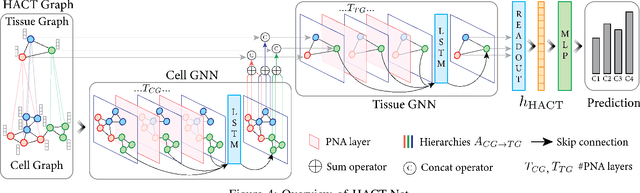
Cancer diagnosis, prognosis, and therapy response predictions from tissue specimens highly depend on the phenotype and topological distribution of constituting histological entities. Thus, adequate tissue representations for encoding histological entities is imperative for computer aided cancer patient care. To this end, several approaches have leveraged cell-graphs that encode cell morphology and organization to denote the tissue information. These allow for utilizing machine learning to map tissue representations to tissue functionality to help quantify their relationship. Though cellular information is crucial, it is incomplete alone to comprehensively characterize complex tissue structure. We herein treat the tissue as a hierarchical composition of multiple types of histological entities from fine to coarse level, capturing multivariate tissue information at multiple levels. We propose a novel multi-level hierarchical entity-graph representation of tissue specimens to model hierarchical compositions that encode histological entities as well as their intra- and inter-entity level interactions. Subsequently, a graph neural network is proposed to operate on the hierarchical entity-graph representation to map the tissue structure to tissue functionality. Specifically, for input histology images we utilize well-defined cells and tissue regions to build HierArchical Cell-to-Tissue (HACT) graph representations, and devise HACT-Net, a graph neural network, to classify such HACT representations. As part of this work, we introduce the BReAst Carcinoma Subtyping (BRACS) dataset, a large cohort of H&E stained breast tumor images, to evaluate our proposed methodology against pathologists and state-of-the-art approaches. Through comparative assessment and ablation studies, our method is demonstrated to yield superior classification results compared to alternative methods as well as pathologists.
Learning Whole-Slide Segmentation from Inexact and Incomplete Labels using Tissue Graphs
Mar 04, 2021Segmenting histology images into diagnostically relevant regions is imperative to support timely and reliable decisions by pathologists. To this end, computer-aided techniques have been proposed to delineate relevant regions in scanned histology slides. However, the techniques necessitate task-specific large datasets of annotated pixels, which is tedious, time-consuming, expensive, and infeasible to acquire for many histology tasks. Thus, weakly-supervised semantic segmentation techniques are proposed to utilize weak supervision that is cheaper and quicker to acquire. In this paper, we propose SegGini, a weakly supervised segmentation method using graphs, that can utilize weak multiplex annotations, i.e. inexact and incomplete annotations, to segment arbitrary and large images, scaling from tissue microarray (TMA) to whole slide image (WSI). Formally, SegGini constructs a tissue-graph representation for an input histology image, where the graph nodes depict tissue regions. Then, it performs weakly-supervised segmentation via node classification by using inexact image-level labels, incomplete scribbles, or both. We evaluated SegGini on two public prostate cancer datasets containing TMAs and WSIs. Our method achieved state-of-the-art segmentation performance on both datasets for various annotation settings while being comparable to a pathologist baseline.
Quantifying Explainers of Graph Neural Networks in Computational Pathology
Nov 25, 2020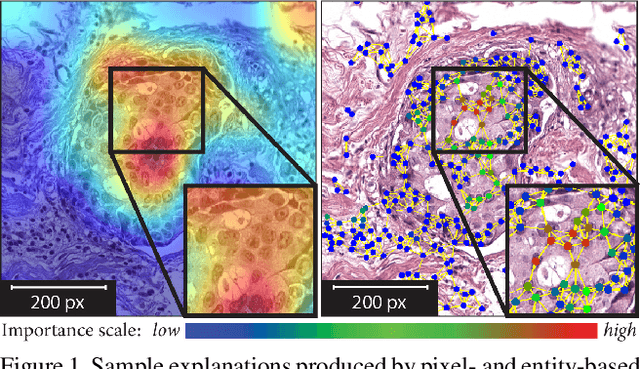
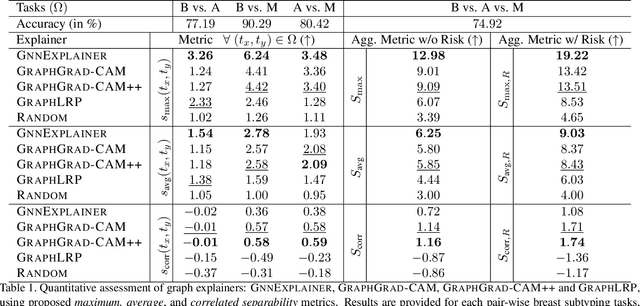
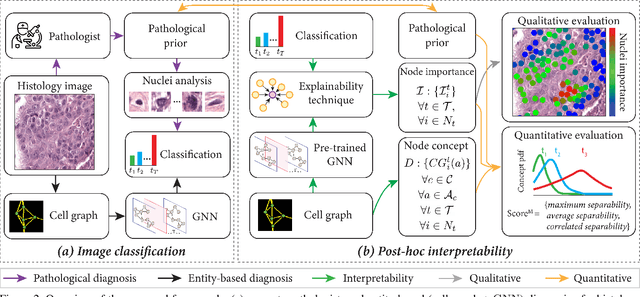
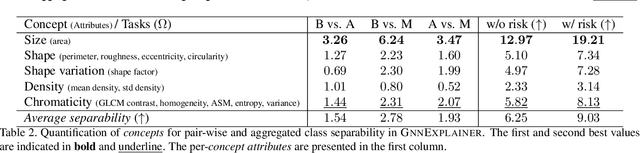
Explainability of deep learning methods is imperative to facilitate their clinical adoption in digital pathology. However, popular deep learning methods and explainability techniques (explainers) based on pixel-wise processing disregard biological entities' notion, thus complicating comprehension by pathologists. In this work, we address this by adopting biological entity-based graph processing and graph explainers enabling explanations accessible to pathologists. In this context, a major challenge becomes to discern meaningful explainers, particularly in a standardized and quantifiable fashion. To this end, we propose herein a set of novel quantitative metrics based on statistics of class separability using pathologically measurable concepts to characterize graph explainers. We employ the proposed metrics to evaluate three types of graph explainers, namely the layer-wise relevance propagation, gradient-based saliency, and graph pruning approaches, to explain Cell-Graph representations for Breast Cancer Subtyping. The proposed metrics are also applicable in other domains by using domain-specific intuitive concepts. We validate the qualitative and quantitative findings on the BRACS dataset, a large cohort of breast cancer RoIs, by expert pathologists.