 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearning 3D Representations of Molecular Chirality with Invariance to Bond Rotations
Oct 08, 2021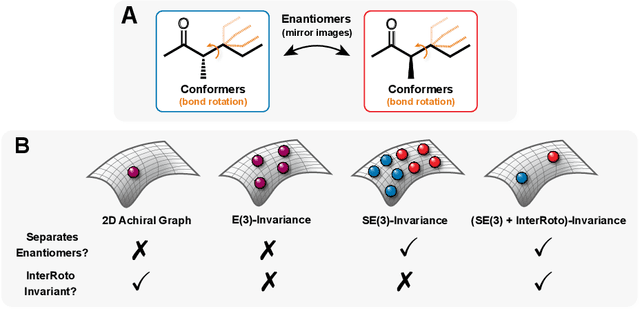

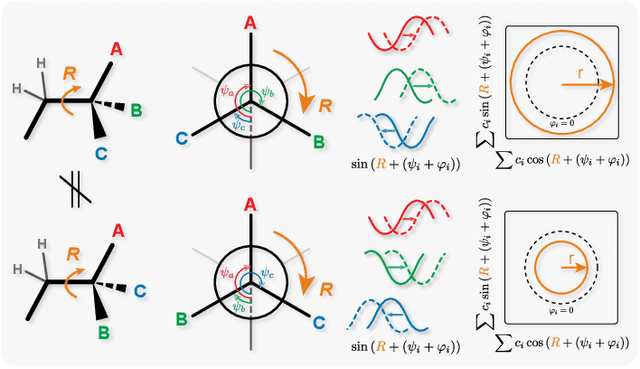
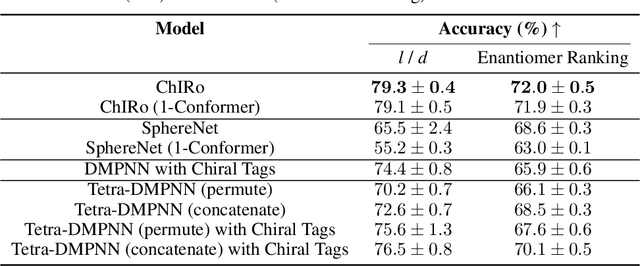
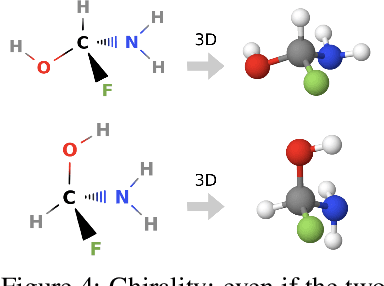
Molecular chirality, a form of stereochemistry most often describing relative spatial arrangements of bonded neighbors around tetrahedral carbon centers, influences the set of 3D conformers accessible to the molecule without changing its 2D graph connectivity. Chirality can strongly alter (bio)chemical interactions, particularly protein-drug binding. Most 2D graph neural networks (GNNs) designed for molecular property prediction at best use atomic labels to na\"ively treat chirality, while E(3)-invariant 3D GNNs are invariant to chirality altogether. To enable representation learning on molecules with defined stereochemistry, we design an SE(3)-invariant model that processes torsion angles of a 3D molecular conformer. We explicitly model conformational flexibility by integrating a novel type of invariance to rotations about internal molecular bonds into the architecture, mitigating the need for multi-conformer data augmentation. We test our model on four benchmarks: contrastive learning to distinguish conformers of different stereoisomers in a learned latent space, classification of chiral centers as R/S, prediction of how enantiomers rotate circularly polarized light, and ranking enantiomers by their docking scores in an enantiosensitive protein pocket. We compare our model, Chiral InterRoto-Invariant Neural Network (ChIRo), with 2D and 3D GNNs to demonstrate that our model achieves state of the art performance when learning chiral-sensitive functions from molecular structures.
Differentiable Scaffolding Tree for Molecular Optimization
Sep 22, 2021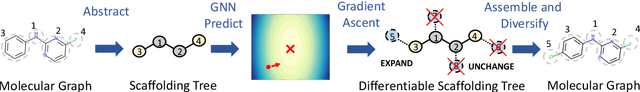

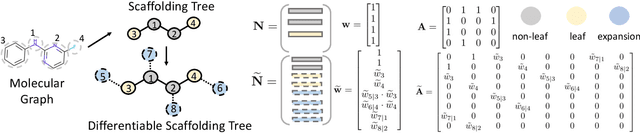
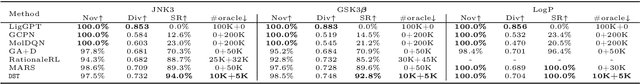
The structural design of functional molecules, also called molecular optimization, is an essential chemical science and engineering task with important applications, such as drug discovery. Deep generative models and combinatorial optimization methods achieve initial success but still struggle with directly modeling discrete chemical structures and often heavily rely on brute-force enumeration. The challenge comes from the discrete and non-differentiable nature of molecule structures. To address this, we propose differentiable scaffolding tree (DST) that utilizes a learned knowledge network to convert discrete chemical structures to locally differentiable ones. DST enables a gradient-based optimization on a chemical graph structure by back-propagating the derivatives from the target properties through a graph neural network (GNN). Our empirical studies show the gradient-based molecular optimizations are both effective and sample efficient. Furthermore, the learned graph parameters can also provide an explanation that helps domain experts understand the model output.
Machine learning modeling of family wide enzyme-substrate specificity screens
Sep 08, 2021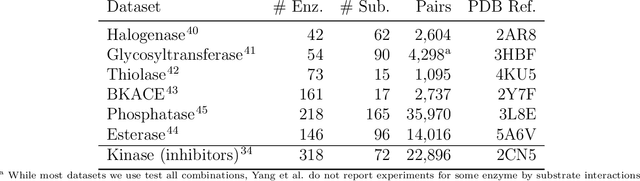
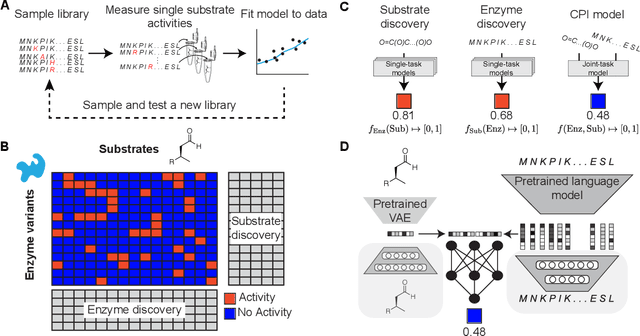
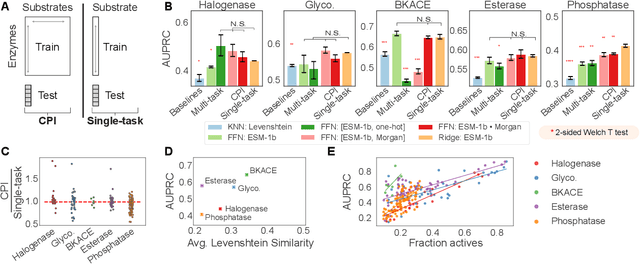
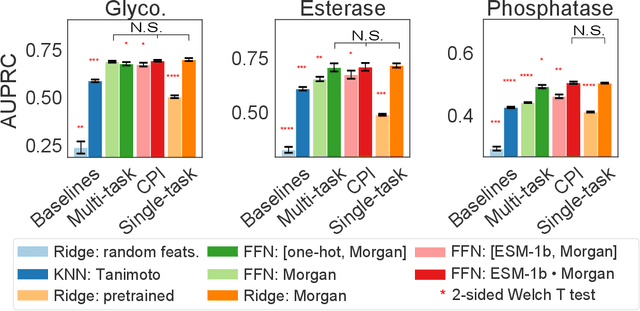
Biocatalysis is a promising approach to sustainably synthesize pharmaceuticals, complex natural products, and commodity chemicals at scale. However, the adoption of biocatalysis is limited by our ability to select enzymes that will catalyze their natural chemical transformation on non-natural substrates. While machine learning and in silico directed evolution are well-posed for this predictive modeling challenge, efforts to date have primarily aimed to increase activity against a single known substrate, rather than to identify enzymes capable of acting on new substrates of interest. To address this need, we curate 6 different high-quality enzyme family screens from the literature that each measure multiple enzymes against multiple substrates. We compare machine learning-based compound-protein interaction (CPI) modeling approaches from the literature used for predicting drug-target interactions. Surprisingly, comparing these interaction-based models against collections of independent (single task) enzyme-only or substrate-only models reveals that current CPI approaches are incapable of learning interactions between compounds and proteins in the current family level data regime. We further validate this observation by demonstrating that our no-interaction baseline can outperform CPI-based models from the literature used to guide the discovery of kinase inhibitors. Given the high performance of non-interaction based models, we introduce a new structure-based strategy for pooling residue representations across a protein sequence. Altogether, this work motivates a principled path forward in order to build and evaluate meaningful predictive models for biocatalysis and other drug discovery applications.
Machine learning on DNA-encoded library count data using an uncertainty-aware probabilistic loss function
Aug 27, 2021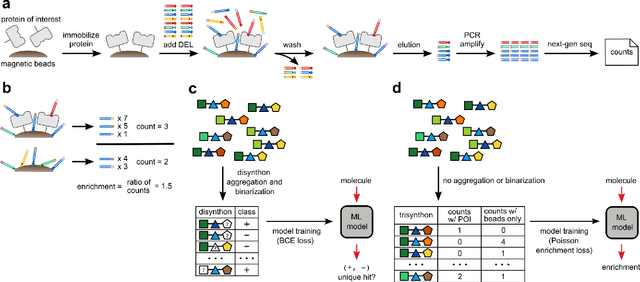
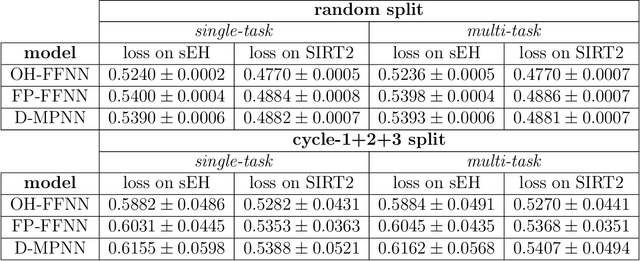
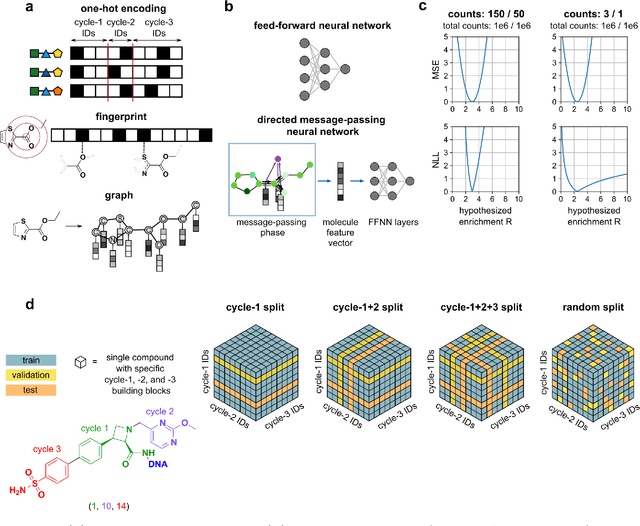
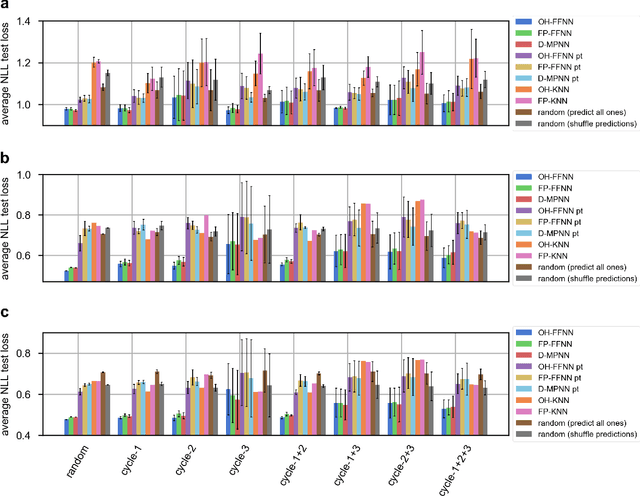
DNA-encoded library (DEL) screening and quantitative structure-activity relationship (QSAR) modeling are two techniques used in drug discovery to find small molecules that bind a protein target. Applying QSAR modeling to DEL data can facilitate the selection of compounds for off-DNA synthesis and evaluation. Such a combined approach has been shown recently by training binary classifiers to learn DEL enrichments of aggregated "disynthons" to accommodate the sparse and noisy nature of DEL data. However, a binary classifier cannot distinguish between different levels of enrichment, and information is potentially lost during disynthon aggregation. Here, we demonstrate a regression approach to learning DEL enrichments of individual molecules using a custom negative log-likelihood loss function that effectively denoises DEL data and introduces opportunities for visualization of learned structure-activity relationships (SAR). Our approach explicitly models the Poisson statistics of the sequencing process used in the DEL experimental workflow under a frequentist view. We illustrate this approach on a dataset of 108k compounds screened against CAIX, and a dataset of 5.7M compounds screened against sEH and SIRT2. Due to the treatment of uncertainty in the data through the negative log-likelihood loss function, the models can ignore low-confidence outliers. While our approach does not demonstrate a benefit for extrapolation to novel structures, we expect our denoising and visualization pipeline to be useful in identifying SAR trends and enriched pharmacophores in DEL data. Further, this approach to uncertainty-aware regression is applicable to other sparse or noisy datasets where the nature of stochasticity is known or can be modeled; in particular, the Poisson enrichment ratio metric we use can apply to other settings that compare sequencing count data between two experimental conditions.
GeoMol: Torsional Geometric Generation of Molecular 3D Conformer Ensembles
Jun 08, 2021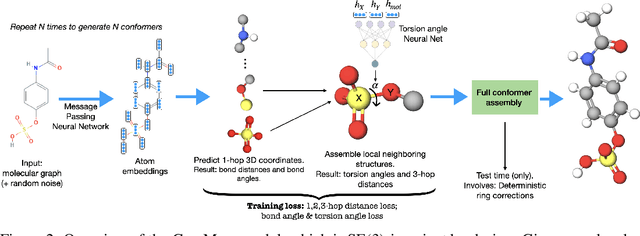
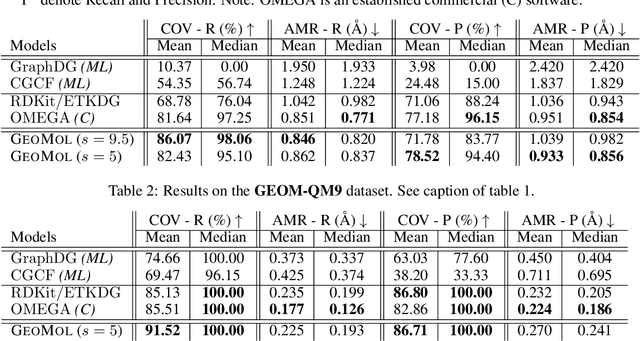
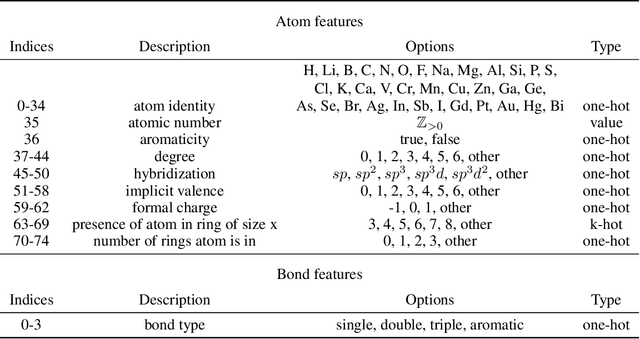
Prediction of a molecule's 3D conformer ensemble from the molecular graph holds a key role in areas of cheminformatics and drug discovery. Existing generative models have several drawbacks including lack of modeling important molecular geometry elements (e.g. torsion angles), separate optimization stages prone to error accumulation, and the need for structure fine-tuning based on approximate classical force-fields or computationally expensive methods such as metadynamics with approximate quantum mechanics calculations at each geometry. We propose GeoMol--an end-to-end, non-autoregressive and SE(3)-invariant machine learning approach to generate distributions of low-energy molecular 3D conformers. Leveraging the power of message passing neural networks (MPNNs) to capture local and global graph information, we predict local atomic 3D structures and torsion angles, avoiding unnecessary over-parameterization of the geometric degrees of freedom (e.g. one angle per non-terminal bond). Such local predictions suffice both for the training loss computation, as well as for the full deterministic conformer assembly (at test time). We devise a non-adversarial optimal transport based loss function to promote diverse conformer generation. GeoMol predominantly outperforms popular open-source, commercial, or state-of-the-art machine learning (ML) models, while achieving significant speed-ups. We expect such differentiable 3D structure generators to significantly impact molecular modeling and related applications.
BioNavi-NP: Biosynthesis Navigator for Natural Products
May 26, 2021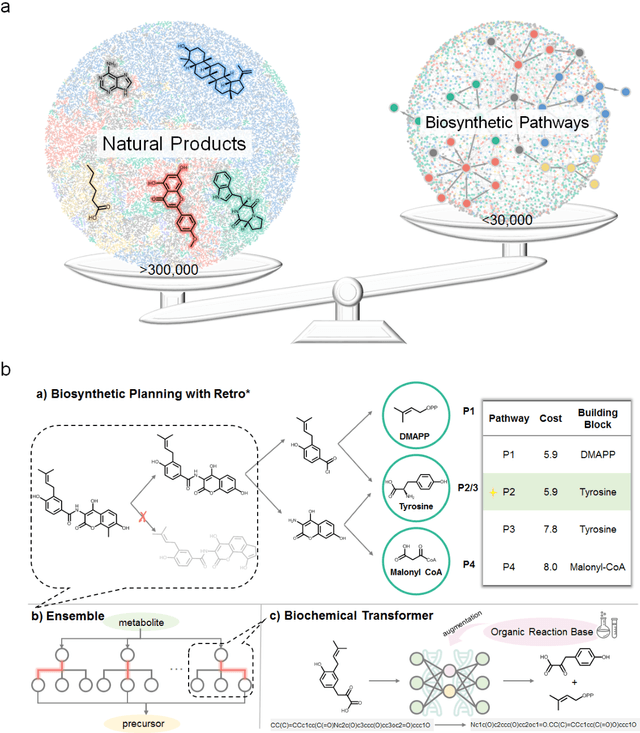
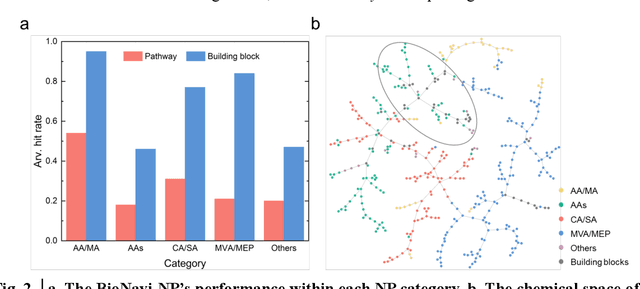
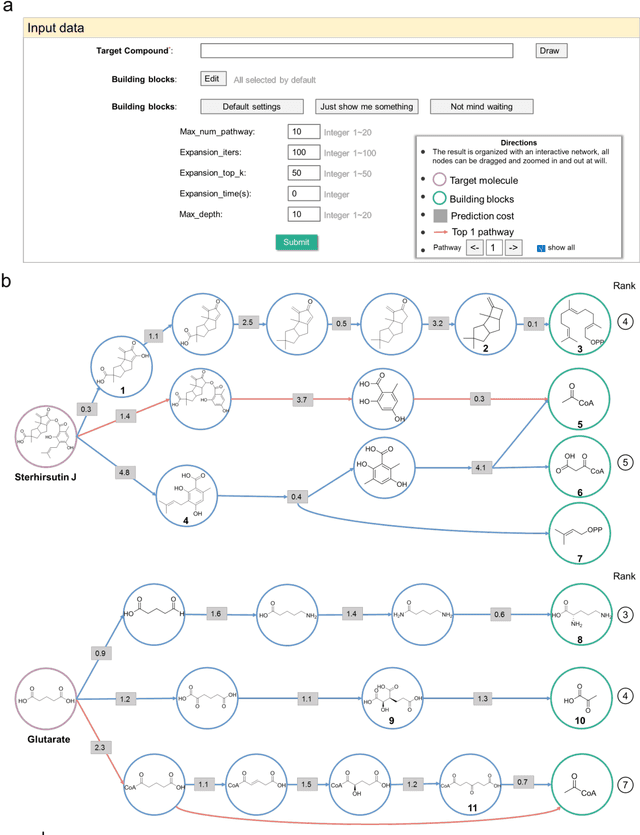
Nature, a synthetic master, creates more than 300,000 natural products (NPs) which are the major constituents of FDA-proved drugs owing to the vast chemical space of NPs. To date, there are fewer than 30,000 validated NPs compounds involved in about 33,000 known enzyme catalytic reactions, and even fewer biosynthetic pathways are known with complete cascade-connected enzyme catalysis. Therefore, it is valuable to make computer-aided bio-retrosynthesis predictions. Here, we develop BioNavi-NP, a navigable and user-friendly toolkit, which is capable of predicting the biosynthetic pathways for NPs and NP-like compounds through a novel (AND-OR Tree)-based planning algorithm, an enhanced molecular Transformer neural network, and a training set that combines general organic transformations and biosynthetic steps. Extensive evaluations reveal that BioNavi-NP generalizes well to identifying the reported biosynthetic pathways for 90% of test compounds and recovering the verified building blocks for 73%, significantly outperforming conventional rule-based approaches. Moreover, BioNavi-NP also shows an outstanding capacity of biologically plausible pathways enumeration. In this sense, BioNavi-NP is a leading-edge toolkit to redesign complex biosynthetic pathways of natural products with applications to total or semi-synthesis and pathway elucidation or reconstruction.
Therapeutics Data Commons: Machine Learning Datasets and Tasks for Therapeutics
Feb 18, 2021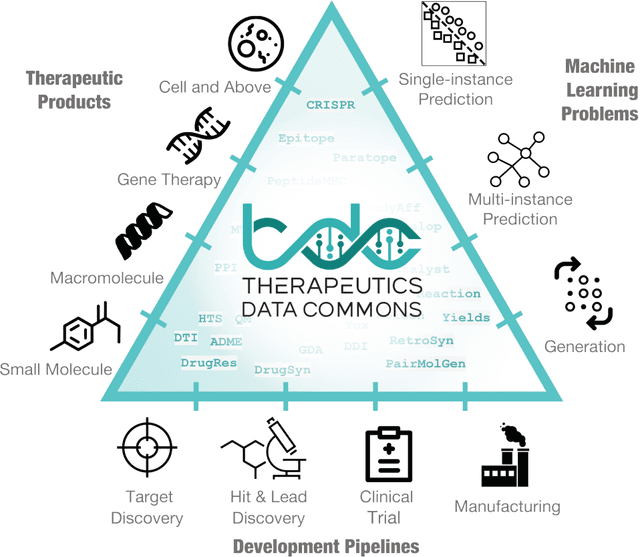
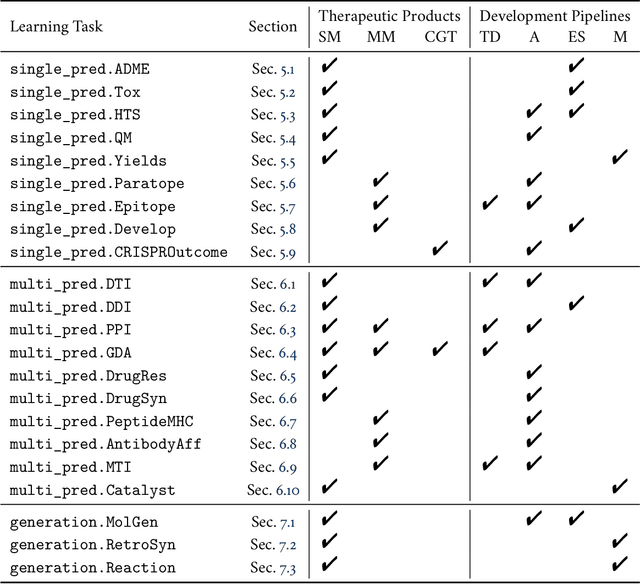
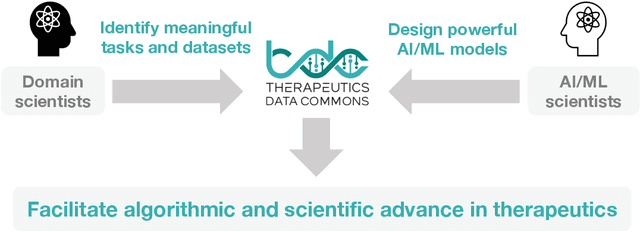
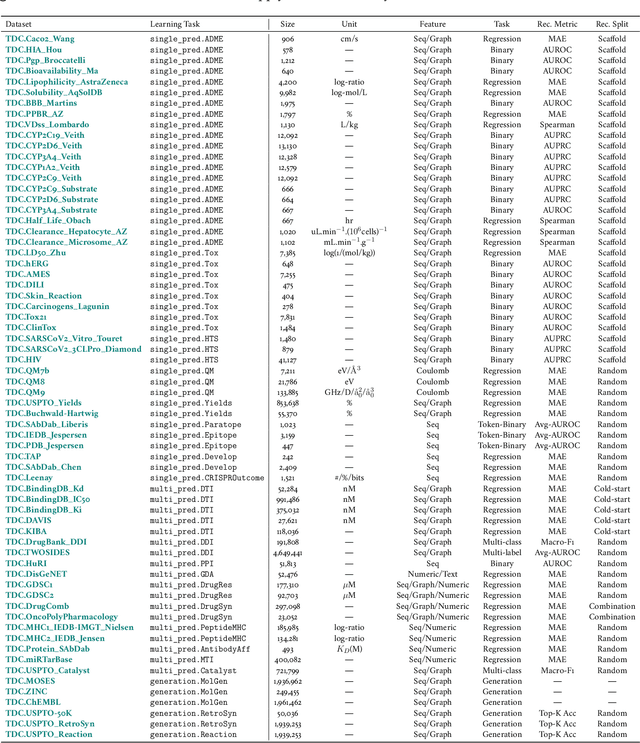
Machine learning for therapeutics is an emerging field with incredible opportunities for innovation and expansion. Despite the initial success, many key challenges remain open. Here, we introduce Therapeutics Data Commons (TDC), the first unifying framework to systematically access and evaluate machine learning across the entire range of therapeutics. At its core, TDC is a collection of curated datasets and learning tasks that can translate algorithmic innovation into biomedical and clinical implementation. To date, TDC includes 66 machine learning-ready datasets from 22 learning tasks, spanning the discovery and development of safe and effective medicines. TDC also provides an ecosystem of tools, libraries, leaderboards, and community resources, including data functions, strategies for systematic model evaluation, meaningful data splits, data processors, and molecule generation oracles. All datasets and learning tasks are integrated and accessible via an open-source library. We envision that TDC can facilitate algorithmic and scientific advances and accelerate development, validation, and transition into production and clinical implementation. TDC is a continuous, open-source initiative, and we invite contributions from the research community. TDC is publicly available at https://tdcommons.ai.
Accelerating high-throughput virtual screening through molecular pool-based active learning
Dec 13, 2020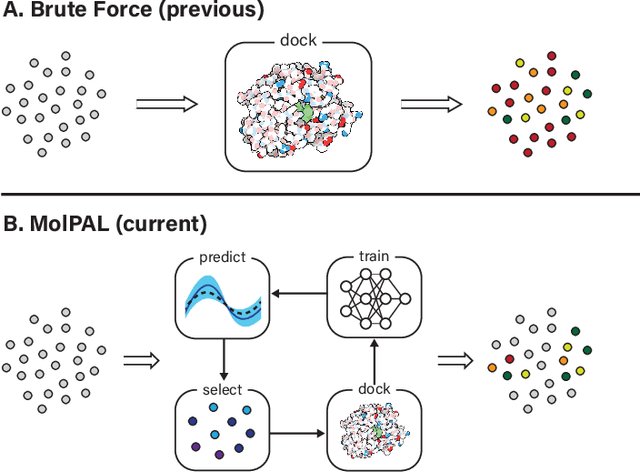
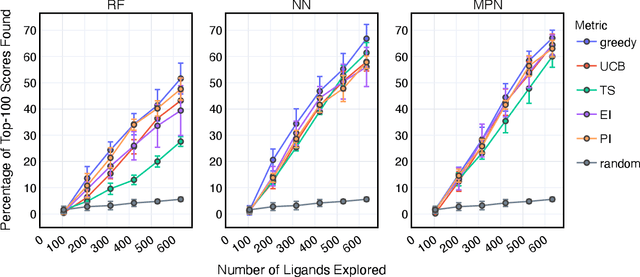
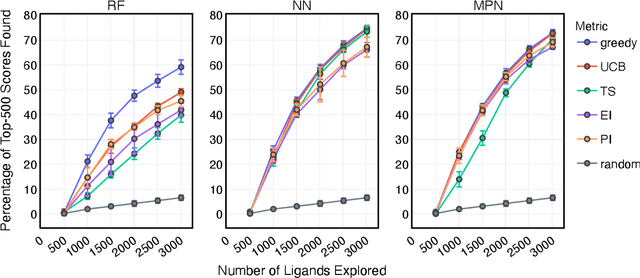
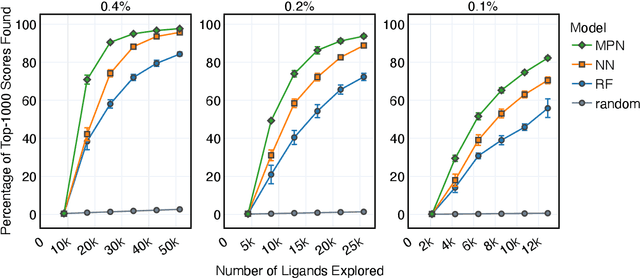
Structure-based virtual screening is an important tool in early stage drug discovery that scores the interactions between a target protein and candidate ligands. As virtual libraries continue to grow (in excess of $10^8$ molecules), so too do the resources necessary to conduct exhaustive virtual screening campaigns on these libraries. However, Bayesian optimization techniques can aid in their exploration: a surrogate structure-property relationship model trained on the predicted affinities of a subset of the library can be applied to the remaining library members, allowing the least promising compounds to be excluded from evaluation. In this study, we assess various surrogate model architectures, acquisition functions, and acquisition batch sizes as applied to several protein-ligand docking datasets and observe significant reductions in computational costs, even when using a greedy acquisition strategy; for example, 87.9% of the top-50000 ligands can be found after testing only 2.4% of a 100M member library. Such model-guided searches mitigate the increasing computational costs of screening increasingly large virtual libraries and can accelerate high-throughput virtual screening campaigns with applications beyond docking.
Message Passing Networks for Molecules with Tetrahedral Chirality
Dec 04, 2020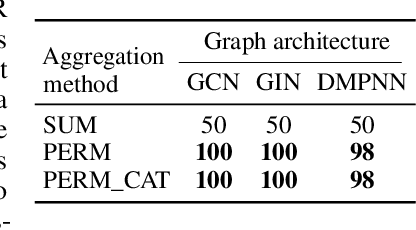
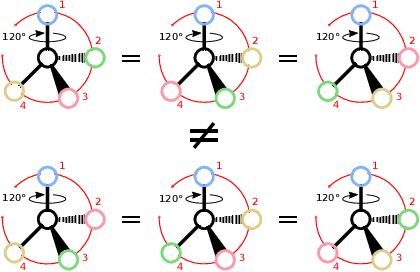
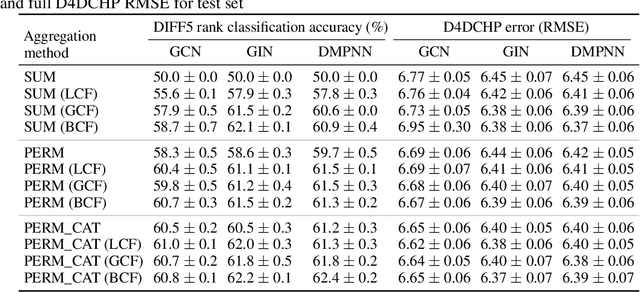
Molecules with identical graph connectivity can exhibit different physical and biological properties if they exhibit stereochemistry-a spatial structural characteristic. However, modern neural architectures designed for learning structure-property relationships from molecular structures treat molecules as graph-structured data and therefore are invariant to stereochemistry. Here, we develop two custom aggregation functions for message passing neural networks to learn properties of molecules with tetrahedral chirality, one common form of stereochemistry. We evaluate performance on synthetic data as well as a newly-proposed protein-ligand docking dataset with relevance to drug discovery. Results show modest improvements over a baseline sum aggregator, highlighting opportunities for further architecture development.
Learning Graph Models for Template-Free Retrosynthesis
Jun 12, 2020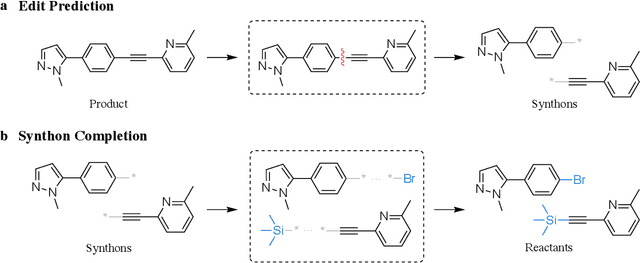
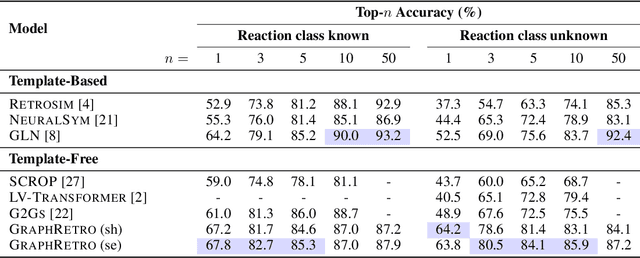
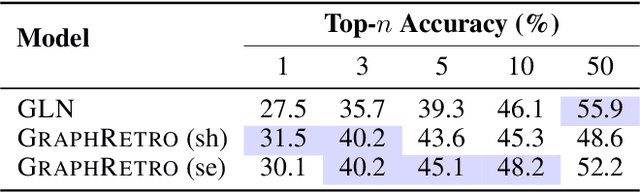
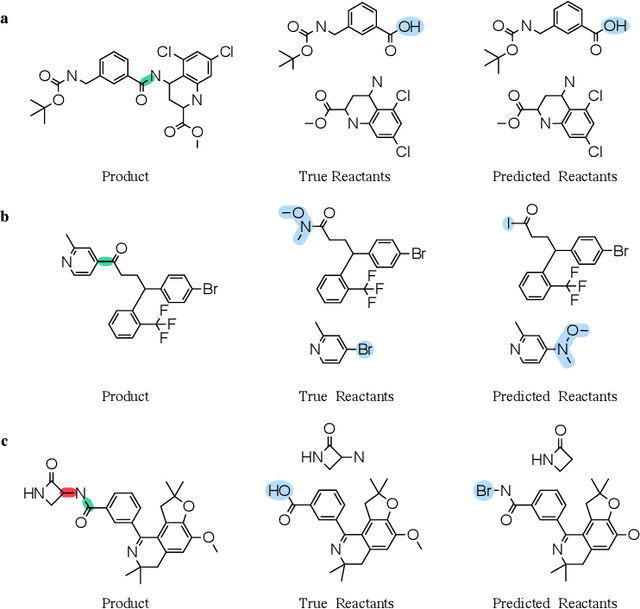
Retrosynthesis prediction is a fundamental problem in organic synthesis, where the task is to identify precursor molecules that can be used to synthesize a target molecule. Despite recent advancements in neural retrosynthesis algorithms, they are unable to fully recapitulate the strategies employed by chemists and do not generalize well to infrequent reaction types. In this paper, we propose a graph-based approach that capitalizes on the idea that the graph topology of precursor molecules is largely unaltered during the reaction. The model first predicts the set of graph edits transforming the target into incomplete molecules called synthons. Next, the model learns to expand synthons into complete molecules by attaching relevant leaving groups. Since the model operates at the level of molecular fragments, it avoids full generation, greatly simplifying the underlying architecture and improving its ability to generalize. The model yields $11.7\%$ absolute improvement over state-of-the-art approaches on the USPTO-50k dataset, and a $4\%$ absolute improvement on a rare reaction subset of the same dataset.