 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMARS: Markov Molecular Sampling for Multi-objective Drug Discovery
Mar 18, 2021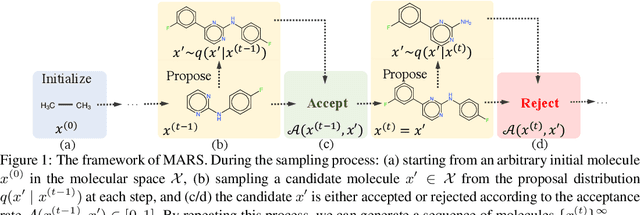
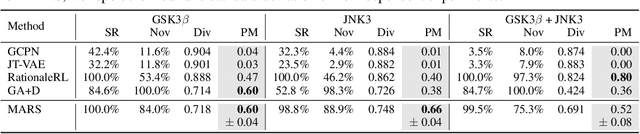
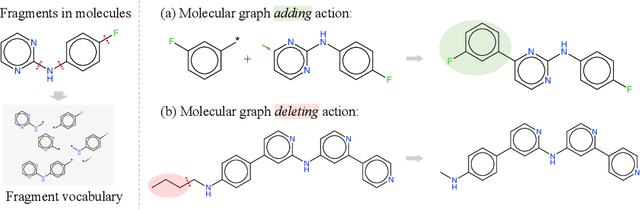
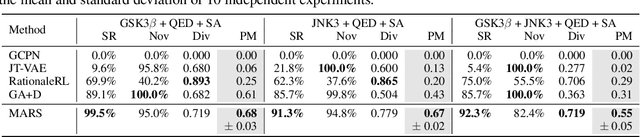
Searching for novel molecules with desired chemical properties is crucial in drug discovery. Existing work focuses on developing neural models to generate either molecular sequences or chemical graphs. However, it remains a big challenge to find novel and diverse compounds satisfying several properties. In this paper, we propose MARS, a method for multi-objective drug molecule discovery. MARS is based on the idea of generating the chemical candidates by iteratively editing fragments of molecular graphs. To search for high-quality candidates, it employs Markov chain Monte Carlo sampling (MCMC) on molecules with an annealing scheme and an adaptive proposal. To further improve sample efficiency, MARS uses a graph neural network (GNN) to represent and select candidate edits, where the GNN is trained on-the-fly with samples from MCMC. Experiments show that MARS achieves state-of-the-art performance in various multi-objective settings where molecular bio-activity, drug-likeness, and synthesizability are considered. Remarkably, in the most challenging setting where all four objectives are simultaneously optimized, our approach outperforms previous methods significantly in comprehensive evaluations. The code is available at https://github.com/yutxie/mars.
Non-autoregressive electron flow generation for reaction prediction
Dec 16, 2020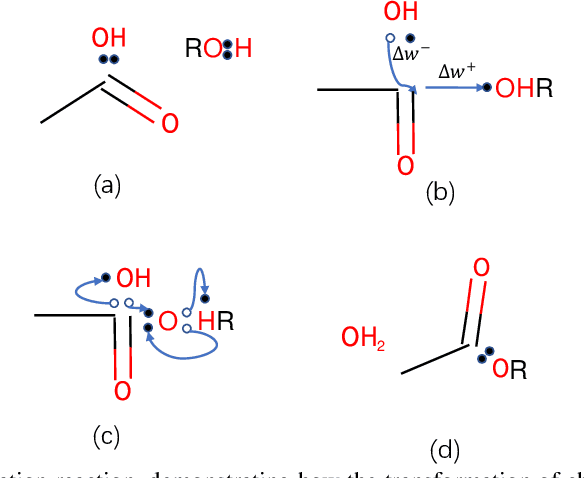
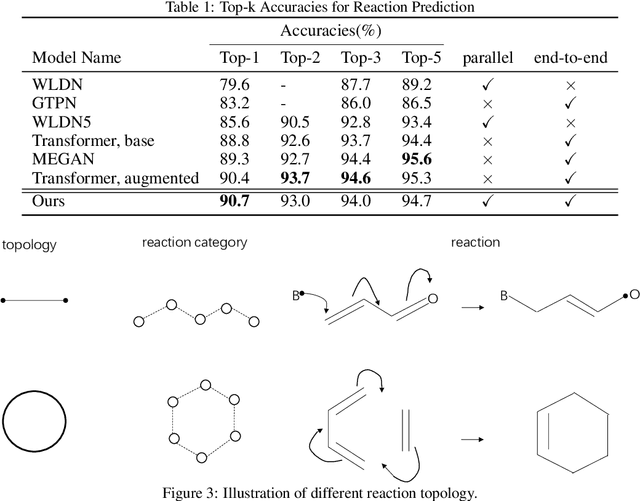
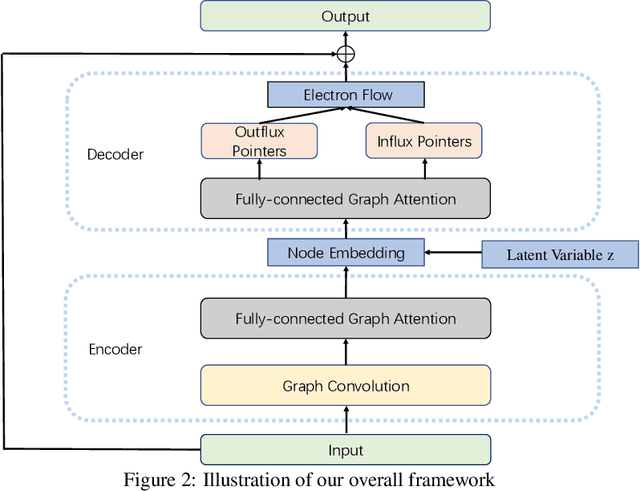

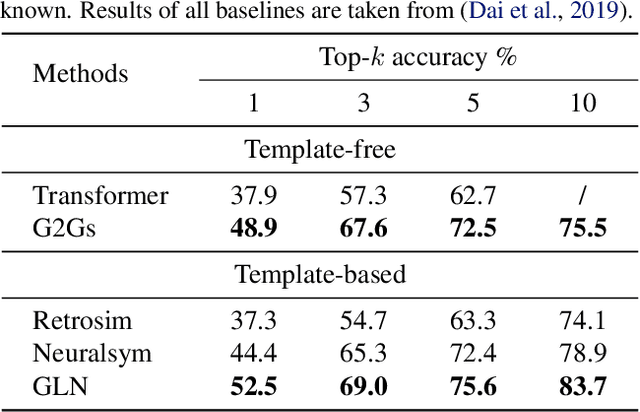
Reaction prediction is a fundamental problem in computational chemistry. Existing approaches typically generate a chemical reaction by sampling tokens or graph edits sequentially, conditioning on previously generated outputs. These autoregressive generating methods impose an arbitrary ordering of outputs and prevent parallel decoding during inference. We devise a novel decoder that avoids such sequential generating and predicts the reaction in a Non-Autoregressive manner. Inspired by physical-chemistry insights, we represent edge edits in a molecule graph as electron flows, which can then be predicted in parallel. To capture the uncertainty of reactions, we introduce latent variables to generate multi-modal outputs. Following previous works, we evaluate our model on USPTO MIT dataset. Our model achieves both an order of magnitude lower inference latency, with state-of-the-art top-1 accuracy and comparable performance on Top-K sampling.
A Graph to Graphs Framework for Retrosynthesis Prediction
Mar 28, 2020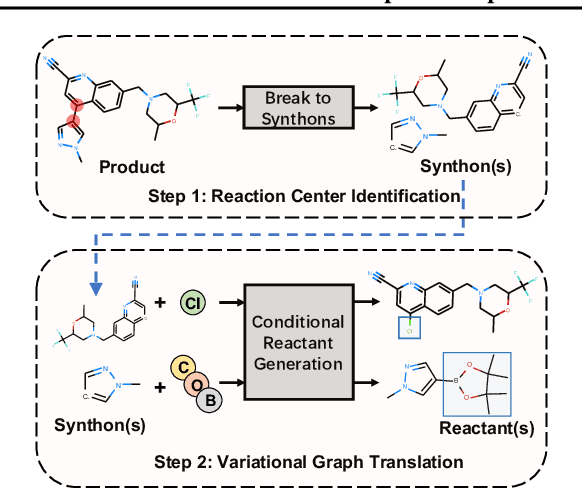
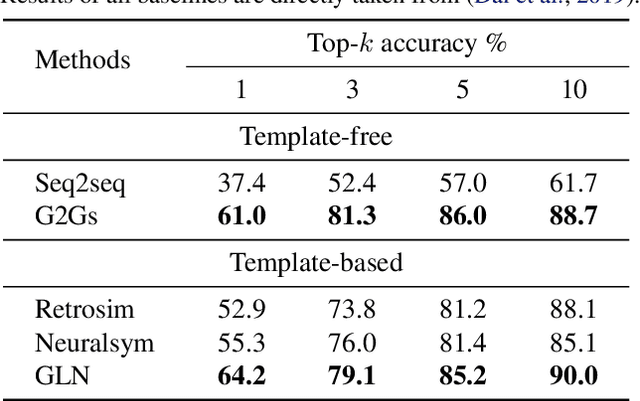
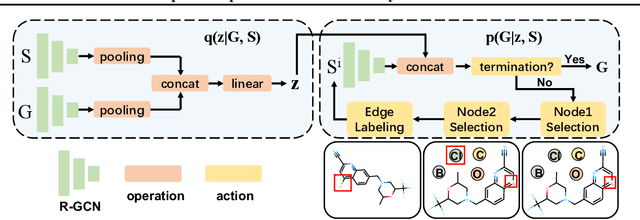
A fundamental problem in computational chemistry is to find a set of reactants to synthesize a target molecule, a.k.a. retrosynthesis prediction. Existing state-of-the-art methods rely on matching the target molecule with a large set of reaction templates, which are very computationally expensive and also suffer from the problem of coverage. In this paper, we propose a novel template-free approach called G2Gs by transforming a target molecular graph into a set of reactant molecular graphs. G2Gs first splits the target molecular graph into a set of synthons by identifying the reaction centers, and then translates the synthons to the final reactant graphs via a variational graph translation framework. Experimental results show that G2Gs significantly outperforms existing template-free approaches by up to 63% in terms of the top-1 accuracy and achieves a performance close to that of state-of-the-art template based approaches, but does not require domain knowledge and is much more scalable.
GraphAF: a Flow-based Autoregressive Model for Molecular Graph Generation
Feb 27, 2020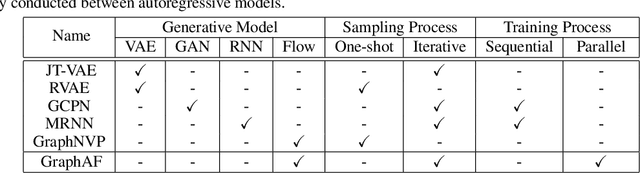
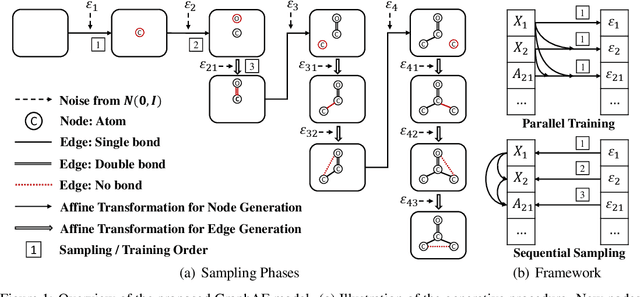
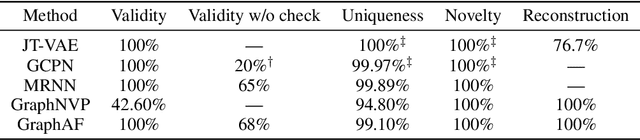
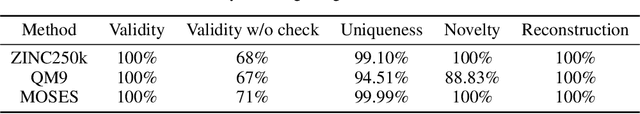
Molecular graph generation is a fundamental problem for drug discovery and has been attracting growing attention. The problem is challenging since it requires not only generating chemically valid molecular structures but also optimizing their chemical properties in the meantime. Inspired by the recent progress in deep generative models, in this paper we propose a flow-based autoregressive model for graph generation called GraphAF. GraphAF combines the advantages of both autoregressive and flow-based approaches and enjoys: (1) high model flexibility for data density estimation; (2) efficient parallel computation for training; (3) an iterative sampling process, which allows leveraging chemical domain knowledge for valency checking. Experimental results show that GraphAF is able to generate 68% chemically valid molecules even without chemical knowledge rules and 100% valid molecules with chemical rules. The training process of GraphAF is two times faster than the existing state-of-the-art approach GCPN. After fine-tuning the model for goal-directed property optimization with reinforcement learning, GraphAF achieves state-of-the-art performance on both chemical property optimization and constrained property optimization.
AutoInt: Automatic Feature Interaction Learning via Self-Attentive Neural Networks
Oct 29, 2018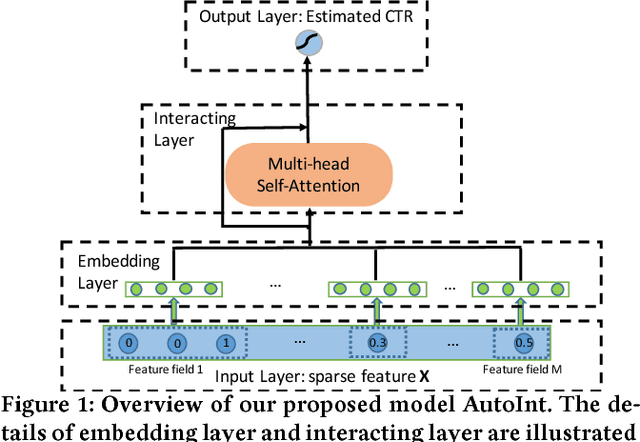
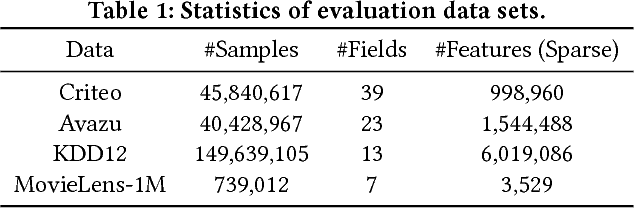
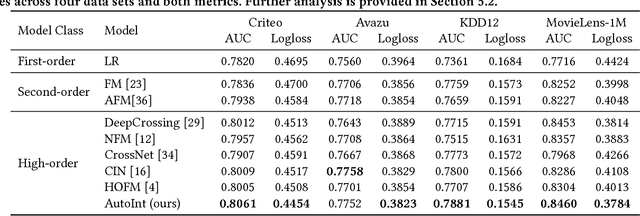
Click-through rate (CTR) prediction, which aims to predict the probability of a user clicking an ad or an item, is critical to many online applications such as online advertising and recommender systems. The problem is very challenging since (1) the input features (e.g., the user id, user age, item id, item category) are usually sparse and high-dimensional, and (2) an effective prediction relies on high-order combinatorial features (a.k.a. cross features), which are very time-consuming to hand-craft by domain experts and are impossible to be enumerated. Therefore, there have been efforts in finding low-dimensional representations of the sparse and high-dimensional raw features and their meaningful combinations. In this paper, we propose an effective and efficient algorithm to automatically learn the high-order feature combinations of input features. Our proposed algorithm is very general, which can be applied to both numerical and categorical input features. Specifically, we map both the numerical and categorical features into the same low-dimensional space. Afterward, a multi-head self-attentive neural network with residual connections is proposed to explicitly model the feature interactions in the low-dimensional space. With different layers of the multi-head self-attentive neural networks, different orders of feature combinations of input features can be modeled. The whole model can be efficiently fit on large-scale raw data in an end-to-end fashion. Experimental results on four real-world datasets show that our proposed approach not only outperforms existing state-of-the-art approaches for prediction but also offers good explainability.