 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVR PreM+ : An Immersive Pre-learning Branching Visualization System for Museum Tours
Nov 01, 2023



We present VR PreM+, an innovative VR system designed to enhance web exploration beyond traditional computer screens. Unlike static 2D displays, VR PreM+ leverages 3D environments to create an immersive pre-learning experience. Using keyword-based information retrieval allows users to manage and connect various content sources in a dynamic 3D space, improving communication and data comparison. We conducted preliminary and user studies that demonstrated efficient information retrieval, increased user engagement, and a greater sense of presence. These findings yielded three design guidelines for future VR information systems: display, interaction, and user-centric design. VR PreM+ bridges the gap between traditional web browsing and immersive VR, offering an interactive and comprehensive approach to information acquisition. It holds promise for research, education, and beyond.
AutoGluon-TimeSeries: AutoML for Probabilistic Time Series Forecasting
Aug 10, 2023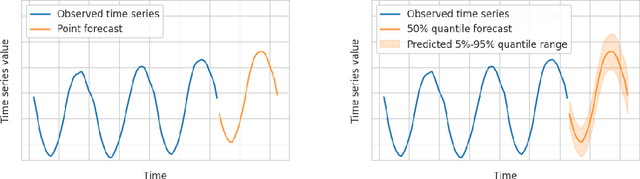
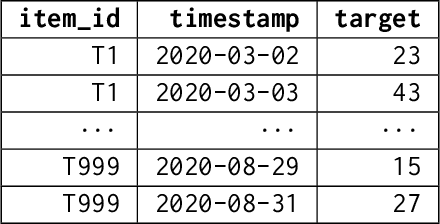
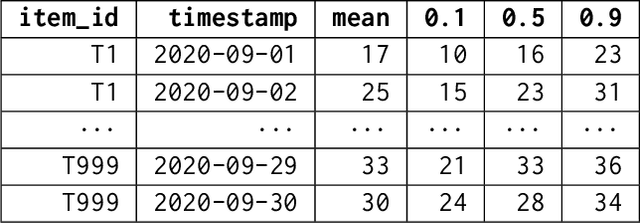
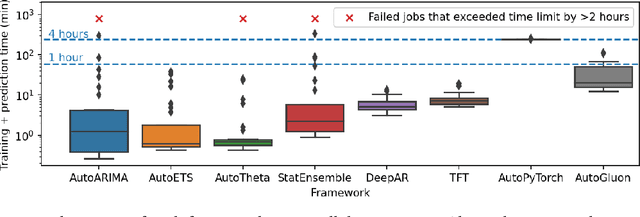
We introduce AutoGluon-TimeSeries - an open-source AutoML library for probabilistic time series forecasting. Focused on ease of use and robustness, AutoGluon-TimeSeries enables users to generate accurate point and quantile forecasts with just 3 lines of Python code. Built on the design philosophy of AutoGluon, AutoGluon-TimeSeries leverages ensembles of diverse forecasting models to deliver high accuracy within a short training time. AutoGluon-TimeSeries combines both conventional statistical models, machine-learning based forecasting approaches, and ensembling techniques. In our evaluation on 29 benchmark datasets, AutoGluon-TimeSeries demonstrates strong empirical performance, outperforming a range of forecasting methods in terms of both point and quantile forecast accuracy, and often even improving upon the best-in-hindsight combination of prior methods.
Predict, Refine, Synthesize: Self-Guiding Diffusion Models for Probabilistic Time Series Forecasting
Jul 21, 2023Diffusion models have achieved state-of-the-art performance in generative modeling tasks across various domains. Prior works on time series diffusion models have primarily focused on developing conditional models tailored to specific forecasting or imputation tasks. In this work, we explore the potential of task-agnostic, unconditional diffusion models for several time series applications. We propose TSDiff, an unconditionally trained diffusion model for time series. Our proposed self-guidance mechanism enables conditioning TSDiff for downstream tasks during inference, without requiring auxiliary networks or altering the training procedure. We demonstrate the effectiveness of our method on three different time series tasks: forecasting, refinement, and synthetic data generation. First, we show that TSDiff is competitive with several task-specific conditional forecasting methods (predict). Second, we leverage the learned implicit probability density of TSDiff to iteratively refine the predictions of base forecasters with reduced computational overhead over reverse diffusion (refine). Notably, the generative performance of the model remains intact -- downstream forecasters trained on synthetic samples from TSDiff outperform forecasters that are trained on samples from other state-of-the-art generative time series models, occasionally even outperforming models trained on real data (synthesize).
PreDiff: Precipitation Nowcasting with Latent Diffusion Models
Jul 19, 2023



Earth system forecasting has traditionally relied on complex physical models that are computationally expensive and require significant domain expertise. In the past decade, the unprecedented increase in spatiotemporal Earth observation data has enabled data-driven forecasting models using deep learning techniques. These models have shown promise for diverse Earth system forecasting tasks but either struggle with handling uncertainty or neglect domain-specific prior knowledge, resulting in averaging possible futures to blurred forecasts or generating physically implausible predictions. To address these limitations, we propose a two-stage pipeline for probabilistic spatiotemporal forecasting: 1) We develop PreDiff, a conditional latent diffusion model capable of probabilistic forecasts. 2) We incorporate an explicit knowledge control mechanism to align forecasts with domain-specific physical constraints. This is achieved by estimating the deviation from imposed constraints at each denoising step and adjusting the transition distribution accordingly. We conduct empirical studies on two datasets: N-body MNIST, a synthetic dataset with chaotic behavior, and SEVIR, a real-world precipitation nowcasting dataset. Specifically, we impose the law of conservation of energy in N-body MNIST and anticipated precipitation intensity in SEVIR. Experiments demonstrate the effectiveness of PreDiff in handling uncertainty, incorporating domain-specific prior knowledge, and generating forecasts that exhibit high operational utility.
Efficient Task Offloading Algorithm for Digital Twin in Edge/Cloud Computing Environment
Jul 13, 2023In the era of Internet of Things (IoT), Digital Twin (DT) is envisioned to empower various areas as a bridge between physical objects and the digital world. Through virtualization and simulation techniques, multiple functions can be achieved by leveraging computing resources. In this process, Mobile Cloud Computing (MCC) and Mobile Edge Computing (MEC) have become two of the key factors to achieve real-time feedback. However, current works only considered edge servers or cloud servers in the DT system models. Besides, The models ignore the DT with not only one data resource. In this paper, we propose a new DT system model considering a heterogeneous MEC/MCC environment. Each DT in the model is maintained in one of the servers via multiple data collection devices. The offloading decision-making problem is also considered and a new offloading scheme is proposed based on Distributed Deep Learning (DDL). Simulation results demonstrate that our proposed algorithm can effectively and efficiently decrease the system's average latency and energy consumption. Significant improvement is achieved compared with the baselines under the dynamic environment of DTs.
Theoretical Guarantees of Learning Ensembling Strategies with Applications to Time Series Forecasting
May 26, 2023



Ensembling is among the most popular tools in machine learning (ML) due to its effectiveness in minimizing variance and thus improving generalization. Most ensembling methods for black-box base learners fall under the umbrella of "stacked generalization," namely training an ML algorithm that takes the inferences from the base learners as input. While stacking has been widely applied in practice, its theoretical properties are poorly understood. In this paper, we prove a novel result, showing that choosing the best stacked generalization from a (finite or finite-dimensional) family of stacked generalizations based on cross-validated performance does not perform "much worse" than the oracle best. Our result strengthens and significantly extends the results in Van der Laan et al. (2007). Inspired by the theoretical analysis, we further propose a particular family of stacked generalizations in the context of probabilistic forecasting, each one with a different sensitivity for how much the ensemble weights are allowed to vary across items, timestamps in the forecast horizon, and quantiles. Experimental results demonstrate the performance gain of the proposed method.
Neural Network Predicts Ion Concentration Profiles under Nanoconfinement
Apr 10, 2023Modeling the ion concentration profile in nanochannel plays an important role in understanding the electrical double layer and electroosmotic flow. Due to the non-negligible surface interaction and the effect of discrete solvent molecules, molecular dynamics (MD) simulation is often used as an essential tool to study the behavior of ions under nanoconfinement. Despite the accuracy of MD simulation in modeling nanoconfinement systems, it is computationally expensive. In this work, we propose neural network to predict ion concentration profiles in nanochannels with different configurations, including channel widths, ion molarity, and ion types. By modeling the ion concentration profile as a probability distribution, our neural network can serve as a much faster surrogate model for MD simulation with high accuracy. We further demonstrate the superior prediction accuracy of neural network over XGBoost. Lastly, we demonstrated that neural network is flexible in predicting ion concentration profiles with different bin sizes. Overall, our deep learning model is a fast, flexible, and accurate surrogate model to predict ion concentration profiles in nanoconfinement.
Testing Causality for High Dimensional Data
Mar 14, 2023
Determining causal relationship between high dimensional observations are among the most important tasks in scientific discoveries. In this paper, we revisited the \emph{linear trace method}, a technique proposed in~\citep{janzing2009telling,zscheischler2011testing} to infer the causal direction between two random variables of high dimensions. We strengthen the existing results significantly by providing an improved tail analysis in addition to extending the results to nonlinear trace functionals with sharper confidence bounds under certain distributional assumptions. We obtain our results by interpreting the trace estimator in the causal regime as a function over random orthogonal matrices, where the concentration of Lipschitz functions over such space could be applied. We additionally propose a novel ridge-regularized variant of the estimator in \cite{zscheischler2011testing}, and give provable bounds relating the ridge-estimated terms to their ground-truth counterparts. We support our theoretical results with encouraging experiments on synthetic datasets, more prominently, under high-dimension low sample size regime.
Denoise Pre-training on Non-equilibrium Molecules for Accurate and Transferable Neural Potentials
Mar 03, 2023Machine learning methods, particularly recent advances in equivariant graph neural networks (GNNs), have been investigated as surrogate models to expensive ab initio quantum mechanics (QM) approaches for molecular potential predictions. However, building accurate and transferable potential models using GNNs remains challenging, as the quality and quantity of data are greatly limited by QM calculations, especially for large and complex molecular systems. In this work, we propose denoise pre-training on non-equilibrium molecular conformations to achieve more accurate and transferable GNN potential predictions. Specifically, GNNs are pre-trained by predicting the random noises added to atomic coordinates of sampled non-equilibrium conformations. Rigorous experiments on multiple benchmarks reveal that pre-training significantly improves the accuracy of neural potentials. Furthermore, we show that the proposed pre-training approach is model-agnostic, as it improves the performance of different invariant and equivariant GNNs. Notably, our models pre-trained on small molecules demonstrate remarkable transferability, improving performance when fine-tuned on diverse molecular systems, including different elements, charged molecules, biomolecules, and larger systems. These results highlight the potential for leveraging denoise pre-training approaches to build more generalizable neural potentials for complex molecular systems.
First De-Trend then Attend: Rethinking Attention for Time-Series Forecasting
Dec 15, 2022



Transformer-based models have gained large popularity and demonstrated promising results in long-term time-series forecasting in recent years. In addition to learning attention in time domain, recent works also explore learning attention in frequency domains (e.g., Fourier domain, wavelet domain), given that seasonal patterns can be better captured in these domains. In this work, we seek to understand the relationships between attention models in different time and frequency domains. Theoretically, we show that attention models in different domains are equivalent under linear conditions (i.e., linear kernel to attention scores). Empirically, we analyze how attention models of different domains show different behaviors through various synthetic experiments with seasonality, trend and noise, with emphasis on the role of softmax operation therein. Both these theoretical and empirical analyses motivate us to propose a new method: TDformer (Trend Decomposition Transformer), that first applies seasonal-trend decomposition, and then additively combines an MLP which predicts the trend component with Fourier attention which predicts the seasonal component to obtain the final prediction. Extensive experiments on benchmark time-series forecasting datasets demonstrate that TDformer achieves state-of-the-art performance against existing attention-based models.