 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReasoning-Driven Design of Single Atom Catalysts via a Multi-Agent Large Language Model Framework
Feb 25, 2026Large language models (LLMs) are becoming increasingly applied beyond natural language processing, demonstrating strong capabilities in complex scientific tasks that traditionally require human expertise. This progress has extended into materials discovery, where LLMs introduce a new paradigm by leveraging reasoning and in-context learning, capabilities absent from conventional machine learning approaches. Here, we present a Multi-Agent-based Electrocatalyst Search Through Reasoning and Optimization (MAESTRO) framework in which multiple LLMs with specialized roles collaboratively discover high-performance single atom catalysts for the oxygen reduction reaction. Within an autonomous design loop, agents iteratively reason, propose modifications, reflect on results and accumulate design history. Through in-context learning enabled by this iterative process, MAESTRO identified design principles not explicitly encoded in the LLMs' background knowledge and successfully discovered catalysts that break conventional scaling relations between reaction intermediates. These results highlight the potential of multi-agent LLM frameworks as a powerful strategy to generate chemical insight and discover promising catalysts.
Facet: highly efficient E(3)-equivariant networks for interatomic potentials
Sep 10, 2025Computational materials discovery is limited by the high cost of first-principles calculations. Machine learning (ML) potentials that predict energies from crystal structures are promising, but existing methods face computational bottlenecks. Steerable graph neural networks (GNNs) encode geometry with spherical harmonics, respecting atomic symmetries -- permutation, rotation, and translation -- for physically realistic predictions. Yet maintaining equivariance is difficult: activation functions must be modified, and each layer must handle multiple data types for different harmonic orders. We present Facet, a GNN architecture for efficient ML potentials, developed through systematic analysis of steerable GNNs. Our innovations include replacing expensive multi-layer perceptrons (MLPs) for interatomic distances with splines, which match performance while cutting computational and memory demands. We also introduce a general-purpose equivariant layer that mixes node information via spherical grid projection followed by standard MLPs -- faster than tensor products and more expressive than linear or gate layers. On the MPTrj dataset, Facet matches leading models with far fewer parameters and under 10% of their training compute. On a crystal relaxation task, it runs twice as fast as MACE models. We further show SevenNet-0's parameters can be reduced by over 25% with no accuracy loss. These techniques enable more than 10x faster training of large-scale foundation models for ML potentials, potentially reshaping computational materials discovery.
MatterTune: An Integrated, User-Friendly Platform for Fine-Tuning Atomistic Foundation Models to Accelerate Materials Simulation and Discovery
Apr 14, 2025



Geometric machine learning models such as graph neural networks have achieved remarkable success in recent years in chemical and materials science research for applications such as high-throughput virtual screening and atomistic simulations. The success of these models can be attributed to their ability to effectively learn latent representations of atomic structures directly from the training data. Conversely, this also results in high data requirements for these models, hindering their application to problems which are data sparse which are common in this domain. To address this limitation, there is a growing development in the area of pre-trained machine learning models which have learned general, fundamental, geometric relationships in atomistic data, and which can then be fine-tuned to much smaller application-specific datasets. In particular, models which are pre-trained on diverse, large-scale atomistic datasets have shown impressive generalizability and flexibility to downstream applications, and are increasingly referred to as atomistic foundation models. To leverage the untapped potential of these foundation models, we introduce MatterTune, a modular and extensible framework that provides advanced fine-tuning capabilities and seamless integration of atomistic foundation models into downstream materials informatics and simulation workflows, thereby lowering the barriers to adoption and facilitating diverse applications in materials science. In its current state, MatterTune supports a number of state-of-the-art foundation models such as ORB, MatterSim, JMP, and EquformerV2, and hosts a wide range of features including a modular and flexible design, distributed and customizable fine-tuning, broad support for downstream informatics tasks, and more.
LLMatDesign: Autonomous Materials Discovery with Large Language Models
Jun 19, 2024



Discovering new materials can have significant scientific and technological implications but remains a challenging problem today due to the enormity of the chemical space. Recent advances in machine learning have enabled data-driven methods to rapidly screen or generate promising materials, but these methods still depend heavily on very large quantities of training data and often lack the flexibility and chemical understanding often desired in materials discovery. We introduce LLMatDesign, a novel language-based framework for interpretable materials design powered by large language models (LLMs). LLMatDesign utilizes LLM agents to translate human instructions, apply modifications to materials, and evaluate outcomes using provided tools. By incorporating self-reflection on its previous decisions, LLMatDesign adapts rapidly to new tasks and conditions in a zero-shot manner. A systematic evaluation of LLMatDesign on several materials design tasks, in silico, validates LLMatDesign's effectiveness in developing new materials with user-defined target properties in the small data regime. Our framework demonstrates the remarkable potential of autonomous LLM-guided materials discovery in the computational setting and towards self-driving laboratories in the future.
On the Quantification of Image Reconstruction Uncertainty without Training Data
Nov 16, 2023



Computational imaging plays a pivotal role in determining hidden information from sparse measurements. A robust inverse solver is crucial to fully characterize the uncertainty induced by these measurements, as it allows for the estimation of the complete posterior of unrecoverable targets. This, in turn, facilitates a probabilistic interpretation of observational data for decision-making. In this study, we propose a deep variational framework that leverages a deep generative model to learn an approximate posterior distribution to effectively quantify image reconstruction uncertainty without the need for training data. We parameterize the target posterior using a flow-based model and minimize their Kullback-Leibler (KL) divergence to achieve accurate uncertainty estimation. To bolster stability, we introduce a robust flow-based model with bi-directional regularization and enhance expressivity through gradient boosting. Additionally, we incorporate a space-filling design to achieve substantial variance reduction on both latent prior space and target posterior space. We validate our method on several benchmark tasks and two real-world applications, namely fastMRI and black hole image reconstruction. Our results indicate that our method provides reliable and high-quality image reconstruction with robust uncertainty estimation.
GDL-DS: A Benchmark for Geometric Deep Learning under Distribution Shifts
Oct 12, 2023



Geometric deep learning (GDL) has gained significant attention in various scientific fields, chiefly for its proficiency in modeling data with intricate geometric structures. Yet, very few works have delved into its capability of tackling the distribution shift problem, a prevalent challenge in many relevant applications. To bridge this gap, we propose GDL-DS, a comprehensive benchmark designed for evaluating the performance of GDL models in scenarios with distribution shifts. Our evaluation datasets cover diverse scientific domains from particle physics and materials science to biochemistry, and encapsulate a broad spectrum of distribution shifts including conditional, covariate, and concept shifts. Furthermore, we study three levels of information access from the out-of-distribution (OOD) testing data, including no OOD information, only OOD features without labels, and OOD features with a few labels. Overall, our benchmark results in 30 different experiment settings, and evaluates 3 GDL backbones and 11 learning algorithms in each setting. A thorough analysis of the evaluation results is provided, poised to illuminate insights for DGL researchers and domain practitioners who are to use DGL in their applications.
Accelerating Inverse Learning via Intelligent Localization with Exploratory Sampling
Dec 02, 2022



In the scope of "AI for Science", solving inverse problems is a longstanding challenge in materials and drug discovery, where the goal is to determine the hidden structures given a set of desirable properties. Deep generative models are recently proposed to solve inverse problems, but these currently use expensive forward operators and struggle in precisely localizing the exact solutions and fully exploring the parameter spaces without missing solutions. In this work, we propose a novel approach (called iPage) to accelerate the inverse learning process by leveraging probabilistic inference from deep invertible models and deterministic optimization via fast gradient descent. Given a target property, the learned invertible model provides a posterior over the parameter space; we identify these posterior samples as an intelligent prior initialization which enables us to narrow down the search space. We then perform gradient descent to calibrate the inverse solutions within a local region. Meanwhile, a space-filling sampling is imposed on the latent space to better explore and capture all possible solutions. We evaluate our approach on three benchmark tasks and two created datasets with real-world applications from quantum chemistry and additive manufacturing, and find our method achieves superior performance compared to several state-of-the-art baseline methods. The iPage code is available at https://github.com/jxzhangjhu/MatDesINNe.
Atomic structure generation from reconstructing structural fingerprints
Jul 27, 2022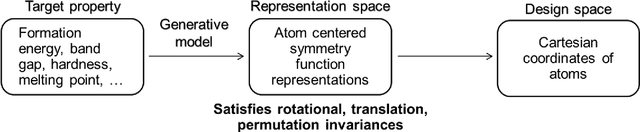
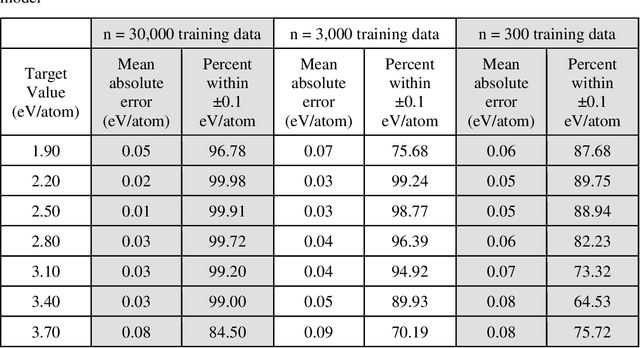
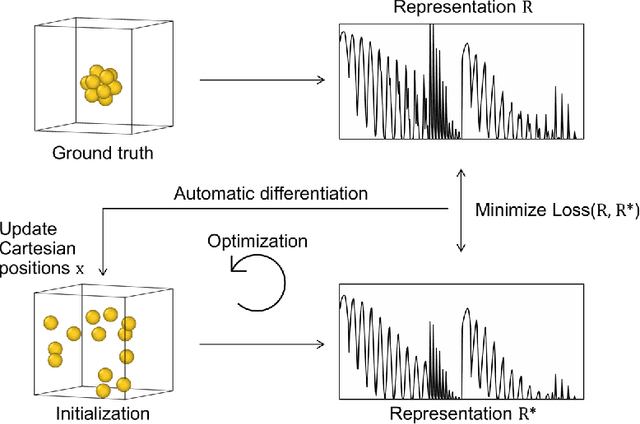
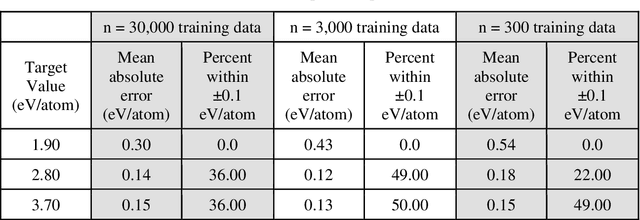
Data-driven machine learning methods have the potential to dramatically accelerate the rate of materials design over conventional human-guided approaches. These methods would help identify or, in the case of generative models, even create novel crystal structures of materials with a set of specified functional properties to then be synthesized or isolated in the laboratory. For crystal structure generation, a key bottleneck lies in developing suitable atomic structure fingerprints or representations for the machine learning model, analogous to the graph-based or SMILES representations used in molecular generation. However, finding data-efficient representations that are invariant to translations, rotations, and permutations, while remaining invertible to the Cartesian atomic coordinates remains an ongoing challenge. Here, we propose an alternative approach to this problem by taking existing non-invertible representations with the desired invariances and developing an algorithm to reconstruct the atomic coordinates through gradient-based optimization using automatic differentiation. This can then be coupled to a generative machine learning model which generates new materials within the representation space, rather than in the data-inefficient Cartesian space. In this work, we implement this end-to-end structure generation approach using atom-centered symmetry functions as the representation and conditional variational autoencoders as the generative model. We are able to successfully generate novel and valid atomic structures of sub-nanometer Pt nanoparticles as a proof of concept. Furthermore, this method can be readily extended to any suitable structural representation, thereby providing a powerful, generalizable framework towards structure-based generation.
Inverse design of two-dimensional materials with invertible neural networks
Jun 06, 2021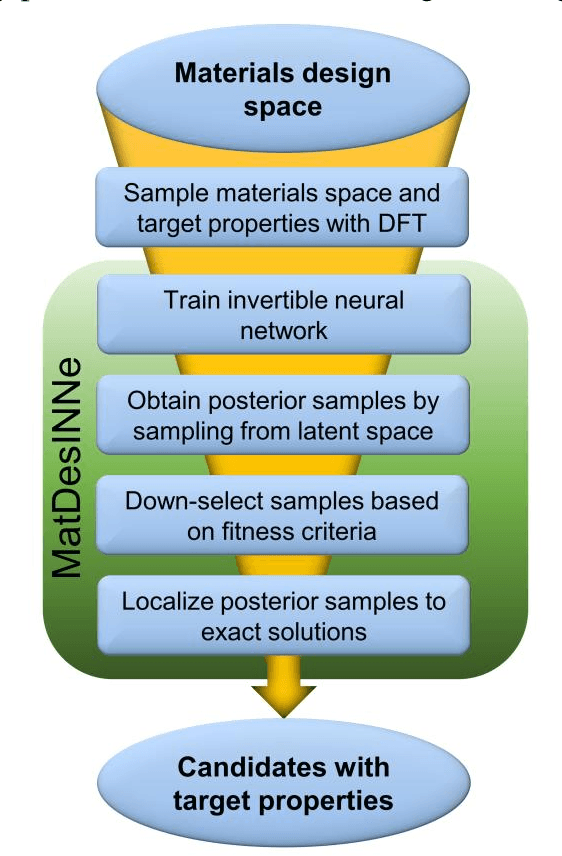
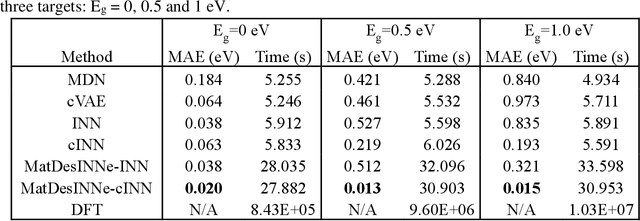
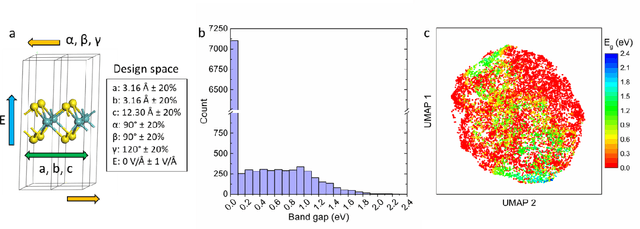
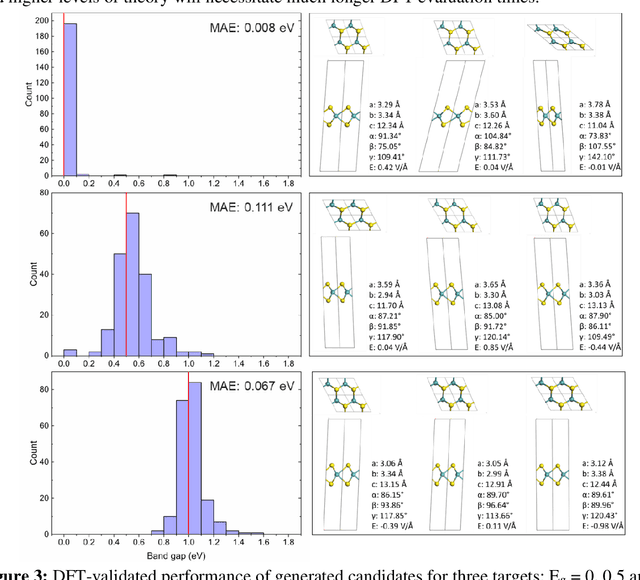
The ability to readily design novel materials with chosen functional properties on-demand represents a next frontier in materials discovery. However, thoroughly and efficiently sampling the entire design space in a computationally tractable manner remains a highly challenging task. To tackle this problem, we propose an inverse design framework (MatDesINNe) utilizing invertible neural networks which can map both forward and reverse processes between the design space and target property. This approach can be used to generate materials candidates for a designated property, thereby satisfying the highly sought-after goal of inverse design. We then apply this framework to the task of band gap engineering in two-dimensional materials, starting with MoS2. Within the design space encompassing six degrees of freedom in applied tensile, compressive and shear strain plus an external electric field, we show the framework can generate novel, high fidelity, and diverse candidates with near-chemical accuracy. We extend this generative capability further to provide insights regarding metal-insulator transition, important for memristive neuromorphic applications among others, in MoS2 which is not otherwise possible with brute force screening. This approach is general and can be directly extended to other materials and their corresponding design spaces and target properties.