 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReasoning-Driven Design of Single Atom Catalysts via a Multi-Agent Large Language Model Framework
Feb 25, 2026Large language models (LLMs) are becoming increasingly applied beyond natural language processing, demonstrating strong capabilities in complex scientific tasks that traditionally require human expertise. This progress has extended into materials discovery, where LLMs introduce a new paradigm by leveraging reasoning and in-context learning, capabilities absent from conventional machine learning approaches. Here, we present a Multi-Agent-based Electrocatalyst Search Through Reasoning and Optimization (MAESTRO) framework in which multiple LLMs with specialized roles collaboratively discover high-performance single atom catalysts for the oxygen reduction reaction. Within an autonomous design loop, agents iteratively reason, propose modifications, reflect on results and accumulate design history. Through in-context learning enabled by this iterative process, MAESTRO identified design principles not explicitly encoded in the LLMs' background knowledge and successfully discovered catalysts that break conventional scaling relations between reaction intermediates. These results highlight the potential of multi-agent LLM frameworks as a powerful strategy to generate chemical insight and discover promising catalysts.
MatterTune: An Integrated, User-Friendly Platform for Fine-Tuning Atomistic Foundation Models to Accelerate Materials Simulation and Discovery
Apr 14, 2025



Geometric machine learning models such as graph neural networks have achieved remarkable success in recent years in chemical and materials science research for applications such as high-throughput virtual screening and atomistic simulations. The success of these models can be attributed to their ability to effectively learn latent representations of atomic structures directly from the training data. Conversely, this also results in high data requirements for these models, hindering their application to problems which are data sparse which are common in this domain. To address this limitation, there is a growing development in the area of pre-trained machine learning models which have learned general, fundamental, geometric relationships in atomistic data, and which can then be fine-tuned to much smaller application-specific datasets. In particular, models which are pre-trained on diverse, large-scale atomistic datasets have shown impressive generalizability and flexibility to downstream applications, and are increasingly referred to as atomistic foundation models. To leverage the untapped potential of these foundation models, we introduce MatterTune, a modular and extensible framework that provides advanced fine-tuning capabilities and seamless integration of atomistic foundation models into downstream materials informatics and simulation workflows, thereby lowering the barriers to adoption and facilitating diverse applications in materials science. In its current state, MatterTune supports a number of state-of-the-art foundation models such as ORB, MatterSim, JMP, and EquformerV2, and hosts a wide range of features including a modular and flexible design, distributed and customizable fine-tuning, broad support for downstream informatics tasks, and more.
Inverse design of two-dimensional materials with invertible neural networks
Jun 06, 2021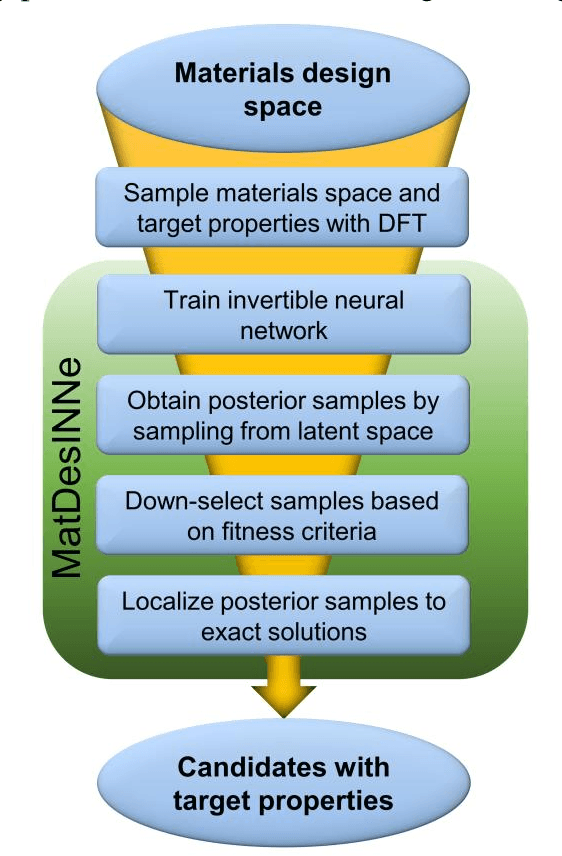
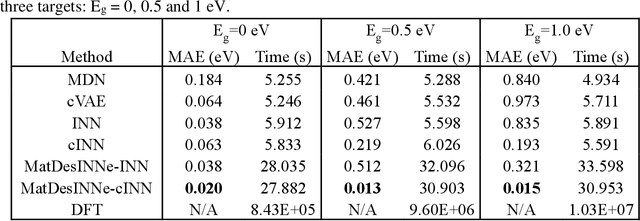
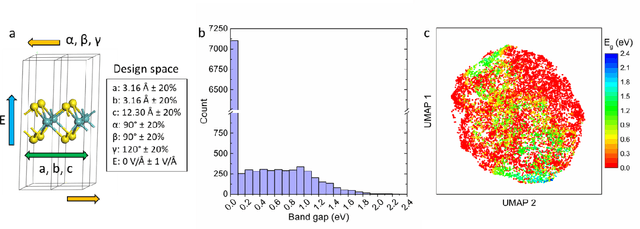
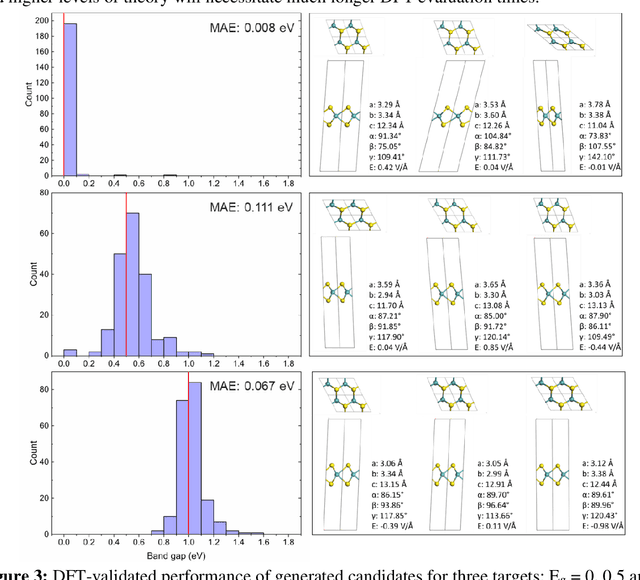
The ability to readily design novel materials with chosen functional properties on-demand represents a next frontier in materials discovery. However, thoroughly and efficiently sampling the entire design space in a computationally tractable manner remains a highly challenging task. To tackle this problem, we propose an inverse design framework (MatDesINNe) utilizing invertible neural networks which can map both forward and reverse processes between the design space and target property. This approach can be used to generate materials candidates for a designated property, thereby satisfying the highly sought-after goal of inverse design. We then apply this framework to the task of band gap engineering in two-dimensional materials, starting with MoS2. Within the design space encompassing six degrees of freedom in applied tensile, compressive and shear strain plus an external electric field, we show the framework can generate novel, high fidelity, and diverse candidates with near-chemical accuracy. We extend this generative capability further to provide insights regarding metal-insulator transition, important for memristive neuromorphic applications among others, in MoS2 which is not otherwise possible with brute force screening. This approach is general and can be directly extended to other materials and their corresponding design spaces and target properties.