 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePyRelationAL: A Library for Active Learning Research and Development
May 23, 2022
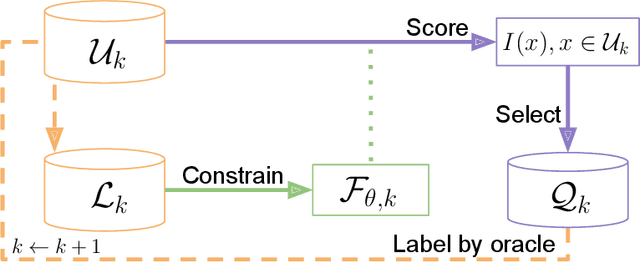
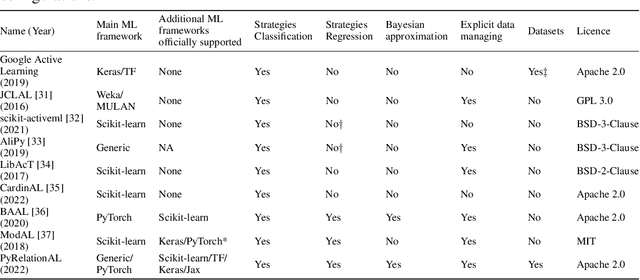
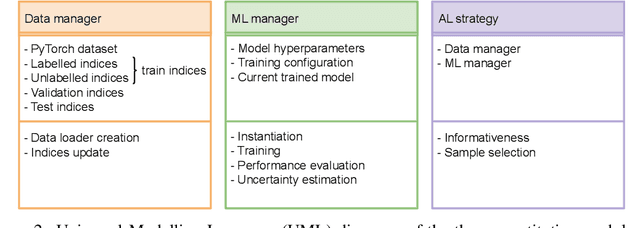
In constrained real-world scenarios where it is challenging or costly to generate data, disciplined methods for acquiring informative new data points are of fundamental importance for the efficient training of machine learning (ML) models. Active learning (AL) is a subfield of ML focused on the development of methods to iteratively and economically acquire data through strategically querying new data points that are the most useful for a particular task. Here, we introduce PyRelationAL, an open source library for AL research. We describe a modular toolkit that is compatible with diverse ML frameworks (e.g. PyTorch, Scikit-Learn, TensorFlow, JAX). Furthermore, to help accelerate research and development in the field, the library implements a number of published methods and provides API access to wide-ranging benchmark datasets and AL task configurations based on existing literature. The library is supplemented by an expansive set of tutorials, demos, and documentation to help users get started. We perform experiments on the PyRelationAL collection of benchmark datasets and showcase the considerable economies that AL can provide. PyRelationAL is maintained using modern software engineering practices - with an inclusive contributor code of conduct - to promote long term library quality and utilisation.
Utilising Graph Machine Learning within Drug Discovery and Development
Dec 09, 2020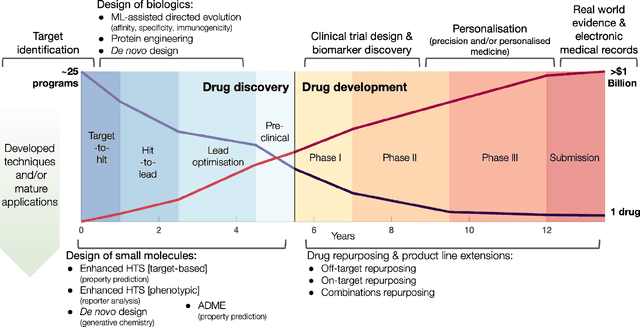
Graph Machine Learning (GML) is receiving growing interest within the pharmaceutical and biotechnology industries for its ability to model biomolecular structures, the functional relationships between them, and integrate multi-omic datasets - amongst other data types. Herein, we present a multidisciplinary academic-industrial review of the topic within the context of drug discovery and development. After introducing key terms and modelling approaches, we move chronologically through the drug development pipeline to identify and summarise work incorporating: target identification, design of small molecules and biologics, and drug repurposing. Whilst the field is still emerging, key milestones including repurposed drugs entering in vivo studies, suggest graph machine learning will become a modelling framework of choice within biomedical machine learning.
Sparse Dynamic Distribution Decomposition: Efficient Integration of Trajectory and Snapshot Time Series Data
Jun 11, 2020
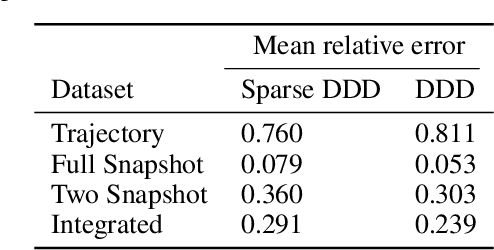
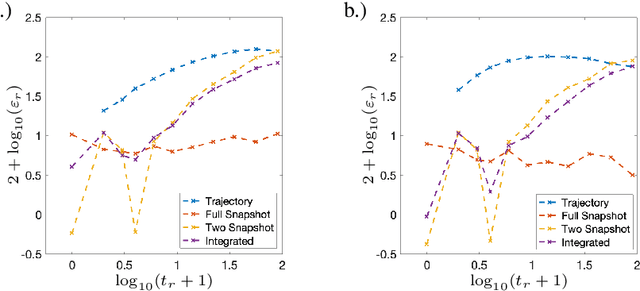
Dynamic Distribution Decomposition (DDD) was introduced in Taylor-King et. al. (PLOS Comp Biol, 2020) as a variation on Dynamic Mode Decomposition. In brief, by using basis functions over a continuous state space, DDD allows for the fitting of continuous-time Markov chains over these basis functions and as a result continuously maps between distributions. The number of parameters in DDD scales by the square of the number of basis functions; we reformulate the problem and restrict the method to compact basis functions which leads to the inference of sparse matrices only -- hence reducing the number of parameters. Finally, we demonstrate how DDD is suitable to integrate both trajectory time series (paired between subsequent time points) and snapshot time series (unpaired time points). Methods capable of integrating both scenarios are particularly relevant for the analysis of biomedical data, whereby studies observe population at fixed time points (snapshots) and individual patient journeys with repeated follow ups (trajectories).