 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOverview of Scanner Invariant Representations
May 29, 2020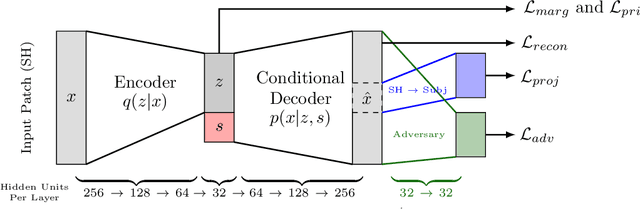
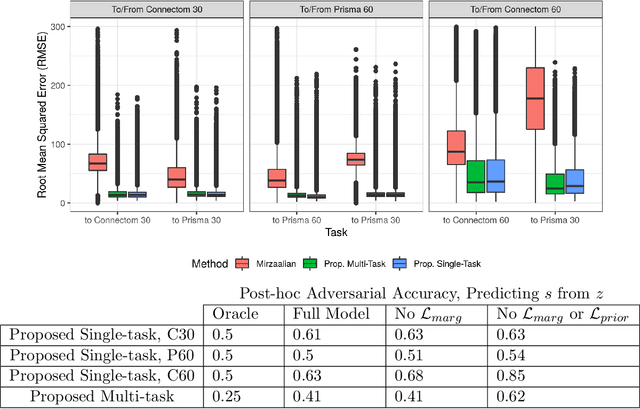
Pooled imaging data from multiple sources is subject to bias from each source. Studies that do not correct for these scanner/site biases at best lose statistical power, and at worst leave spurious correlations in their data. Estimation of the bias effects is non-trivial due to the paucity of data with correspondence across sites, so called "traveling phantom" data, which is expensive to collect. Nevertheless, numerous solutions leveraging direct correspondence have been proposed. In contrast to this, Moyer et al. (2019) proposes an unsupervised solution using invariant representations, one which does not require correspondence and thus does not require paired images. By leveraging the data processing inequality, an invariant representation can then be used to create an image reconstruction that is uninformative of its original source, yet still faithful to the underlying structure. In the present abstract we provide an overview of this method.
Scanner Invariant Representations for Diffusion MRI Harmonization
Apr 10, 2019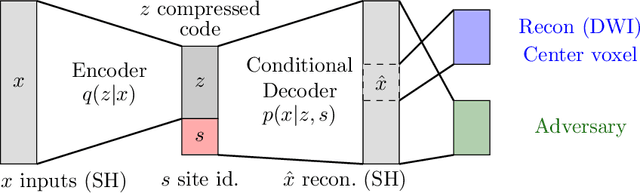
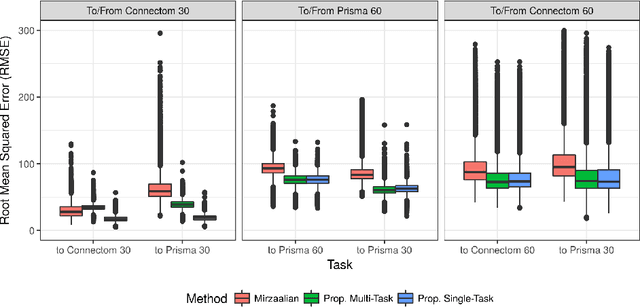
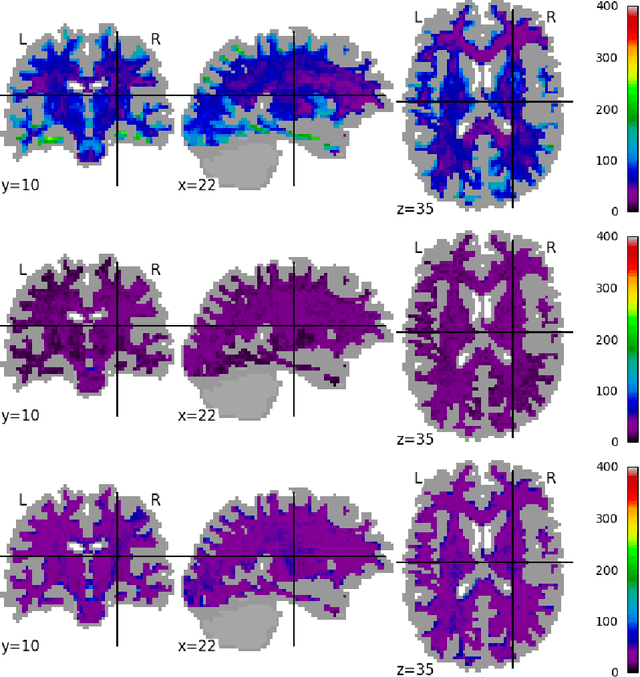
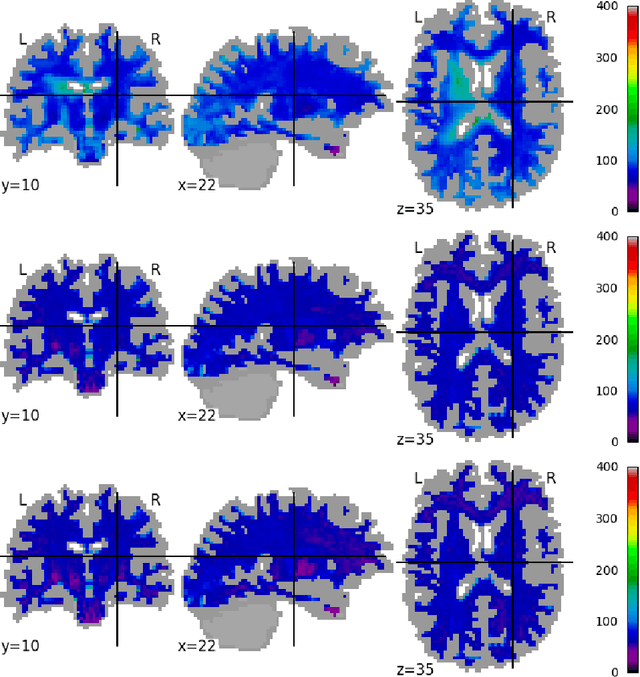
Pooled imaging data from multiple sources is subject to variation between the sources. Correcting for these biases has become incredibly important as the size of imaging studies increases and the multi-site case becomes more common. We propose learning an intermediate representation invariant to site/protocol variables, a technique adapted from information theory-based algorithmic fairness; by leveraging the data processing inequality, such a representation can then be used to create an image reconstruction that is uninformative of its original source, yet still faithful to the underlying structure. To implement this, we use a machine learning method based on variational auto-encoders (VAE) to construct scanner invariant encodings of the imaging data. To evaluate our method, we use training data from the 2018 CDMRI Challenge Harmonization dataset. Our proposed method shows improvements on independent test data relative to a recently published baseline method.
Measures of Tractography Convergence
Jun 12, 2018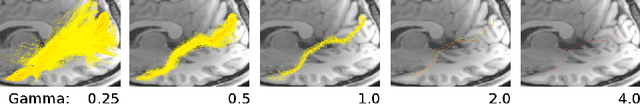

In the present work, we use information theory to understand the empirical convergence rate of tractography, a widely-used approach to reconstruct anatomical fiber pathways in the living brain. Based on diffusion MRI data, tractography is the starting point for many methods to study brain connectivity. Of the available methods to perform tractography, most reconstruct a finite set of streamlines, or 3D curves, representing probable connections between anatomical regions, yet relatively little is known about how the sampling of this set of streamlines affects downstream results, and how exhaustive the sampling should be. Here we provide a method to measure the information theoretic surprise (self-cross entropy) for tract sampling schema. We then empirically assess four streamline methods. We demonstrate that the relative information gain is very low after a moderate number of streamlines have been generated for each tested method. The results give rise to several guidelines for optimal sampling in brain connectivity analyses.
Simultaneous Matrix Diagonalization for Structural Brain Networks Classification
Oct 14, 2017
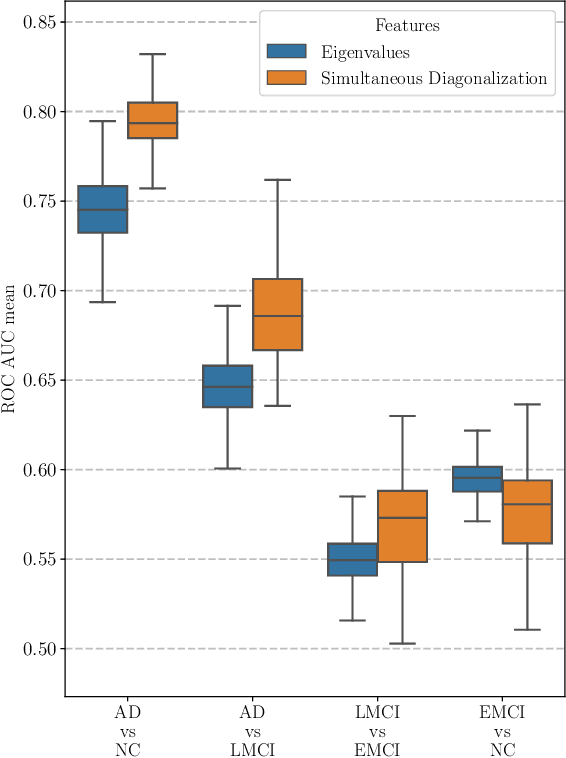

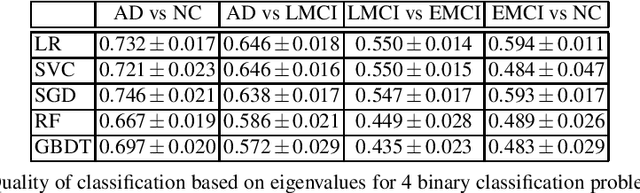
This paper considers the problem of brain disease classification based on connectome data. A connectome is a network representation of a human brain. The typical connectome classification problem is very challenging because of the small sample size and high dimensionality of the data. We propose to use simultaneous approximate diagonalization of adjacency matrices in order to compute their eigenstructures in more stable way. The obtained approximate eigenvalues are further used as features for classification. The proposed approach is demonstrated to be efficient for detection of Alzheimer's disease, outperforming simple baselines and competing with state-of-the-art approaches to brain disease classification.
Classification of Major Depressive Disorder via Multi-Site Weighted LASSO Model
Jun 03, 2017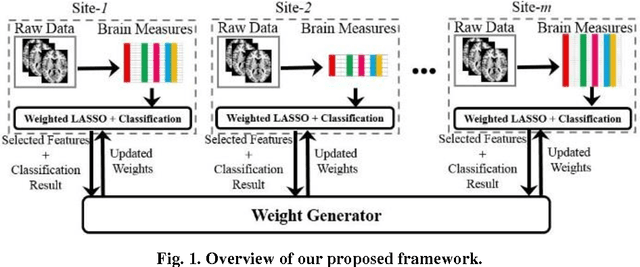
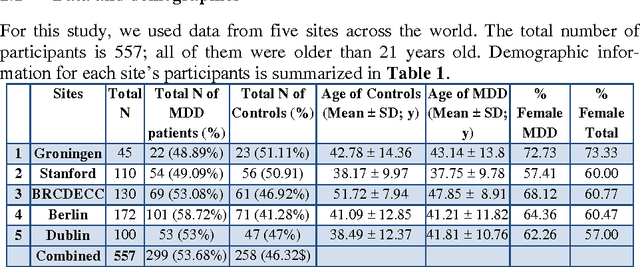

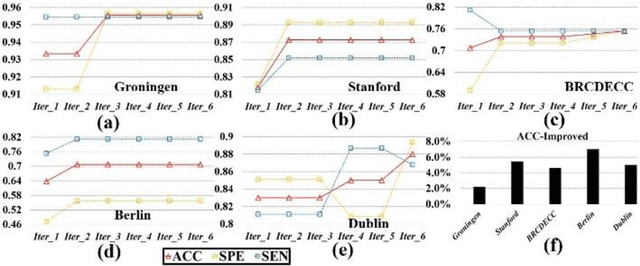
Large-scale collaborative analysis of brain imaging data, in psychiatry and neu-rology, offers a new source of statistical power to discover features that boost ac-curacy in disease classification, differential diagnosis, and outcome prediction. However, due to data privacy regulations or limited accessibility to large datasets across the world, it is challenging to efficiently integrate distributed information. Here we propose a novel classification framework through multi-site weighted LASSO: each site performs an iterative weighted LASSO for feature selection separately. Within each iteration, the classification result and the selected features are collected to update the weighting parameters for each feature. This new weight is used to guide the LASSO process at the next iteration. Only the fea-tures that help to improve the classification accuracy are preserved. In tests on da-ta from five sites (299 patients with major depressive disorder (MDD) and 258 normal controls), our method boosted classification accuracy for MDD by 4.9% on average. This result shows the potential of the proposed new strategy as an ef-fective and practical collaborative platform for machine learning on large scale distributed imaging and biobank data.
Large-scale Feature Selection of Risk Genetic Factors for Alzheimer's Disease via Distributed Group Lasso Regression
Apr 27, 2017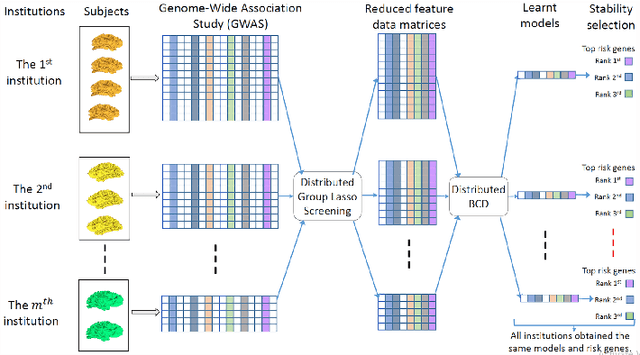
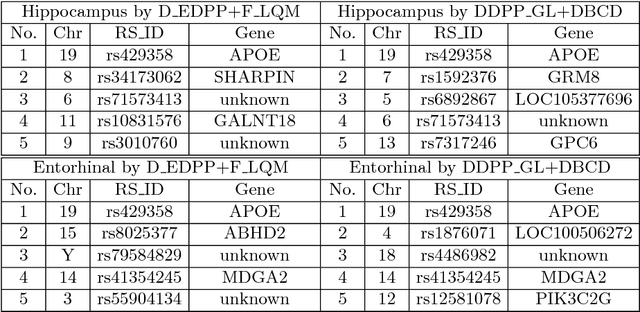
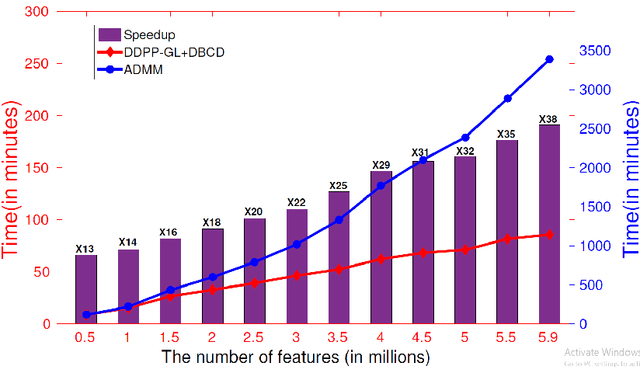
Genome-wide association studies (GWAS) have achieved great success in the genetic study of Alzheimer's disease (AD). Collaborative imaging genetics studies across different research institutions show the effectiveness of detecting genetic risk factors. However, the high dimensionality of GWAS data poses significant challenges in detecting risk SNPs for AD. Selecting relevant features is crucial in predicting the response variable. In this study, we propose a novel Distributed Feature Selection Framework (DFSF) to conduct the large-scale imaging genetics studies across multiple institutions. To speed up the learning process, we propose a family of distributed group Lasso screening rules to identify irrelevant features and remove them from the optimization. Then we select the relevant group features by performing the group Lasso feature selection process in a sequence of parameters. Finally, we employ the stability selection to rank the top risk SNPs that might help detect the early stage of AD. To the best of our knowledge, this is the first distributed feature selection model integrated with group Lasso feature selection as well as detecting the risk genetic factors across multiple research institutions system. Empirical studies are conducted on 809 subjects with 5.9 million SNPs which are distributed across several individual institutions, demonstrating the efficiency and effectiveness of the proposed method.
A Restaurant Process Mixture Model for Connectivity Based Parcellation of the Cortex
Mar 02, 2017
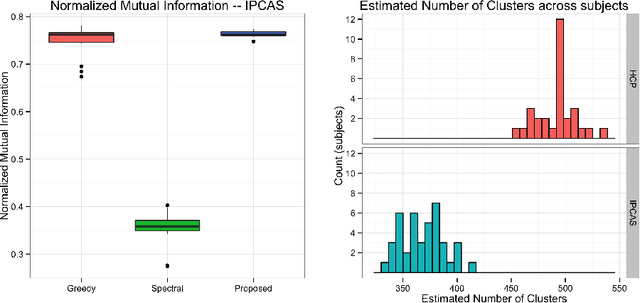
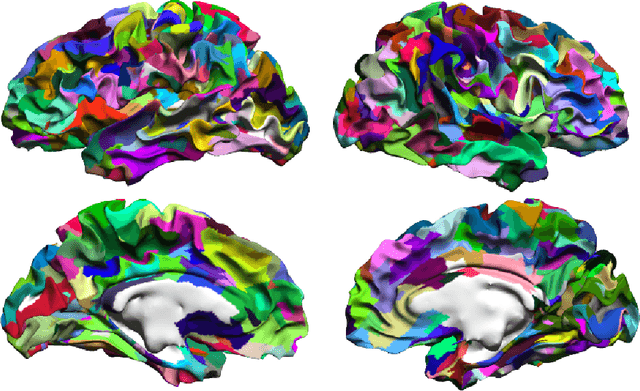
One of the primary objectives of human brain mapping is the division of the cortical surface into functionally distinct regions, i.e. parcellation. While it is generally agreed that at macro-scale different regions of the cortex have different functions, the exact number and configuration of these regions is not known. Methods for the discovery of these regions are thus important, particularly as the volume of available information grows. Towards this end, we present a parcellation method based on a Bayesian non-parametric mixture model of cortical connectivity.
An Empirical Study of Continuous Connectivity Degree Sequence Equivalents
Nov 18, 2016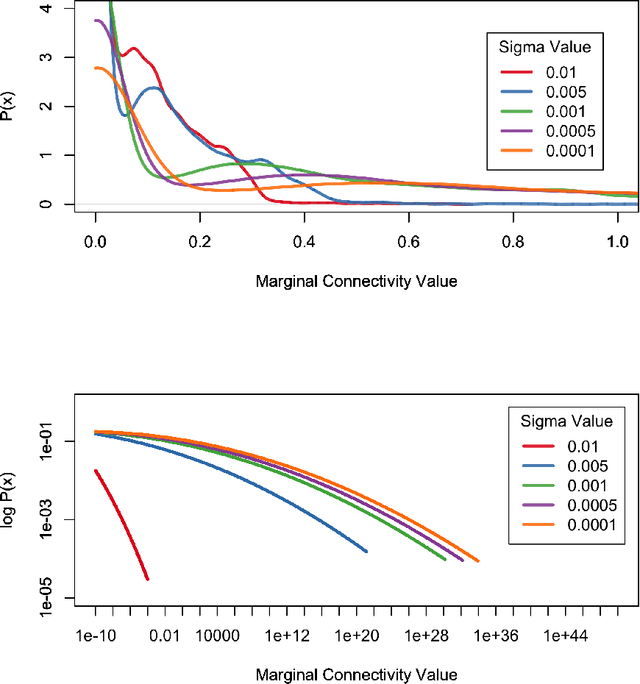

In the present work we demonstrate the use of a parcellation free connectivity model based on Poisson point processes. This model produces for each subject a continuous bivariate intensity function that represents for every possible pair of points the relative rate at which we observe tracts terminating at those points. We fit this model to explore degree sequence equivalents for spatial continuum graphs, and to investigate the local differences between estimated intensity functions for two different tractography methods. This is a companion paper to Moyer et al. (2016), where the model was originally defined.
Large-scale Collaborative Imaging Genetics Studies of Risk Genetic Factors for Alzheimer's Disease Across Multiple Institutions
Aug 19, 2016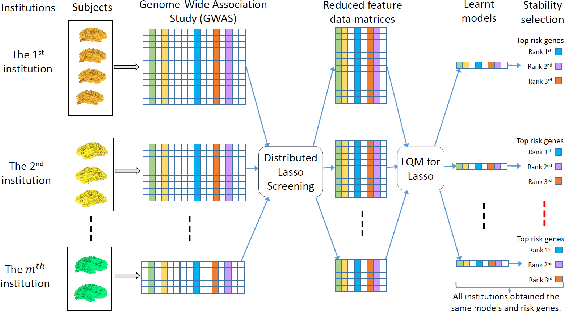
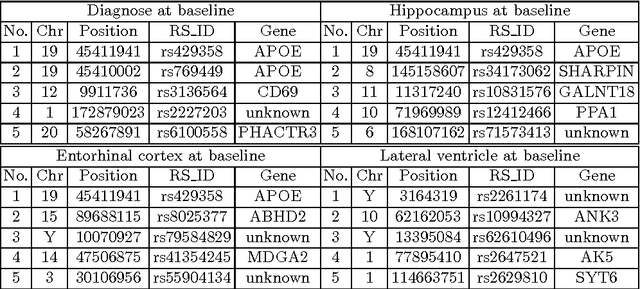
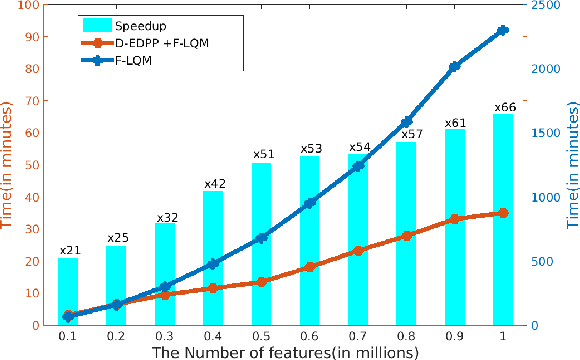
Genome-wide association studies (GWAS) offer new opportunities to identify genetic risk factors for Alzheimer's disease (AD). Recently, collaborative efforts across different institutions emerged that enhance the power of many existing techniques on individual institution data. However, a major barrier to collaborative studies of GWAS is that many institutions need to preserve individual data privacy. To address this challenge, we propose a novel distributed framework, termed Local Query Model (LQM) to detect risk SNPs for AD across multiple research institutions. To accelerate the learning process, we propose a Distributed Enhanced Dual Polytope Projection (D-EDPP) screening rule to identify irrelevant features and remove them from the optimization. To the best of our knowledge, this is the first successful run of the computationally intensive model selection procedure to learn a consistent model across different institutions without compromising their privacy while ranking the SNPs that may collectively affect AD. Empirical studies are conducted on 809 subjects with 5.9 million SNP features which are distributed across three individual institutions. D-EDPP achieved a 66-fold speed-up by effectively identifying irrelevant features.