 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGOLDMARK: Governed Outcome-Linked Diagnostic Model Assessment Reference Kit
Mar 21, 2026Computational biomarkers (CBs) are histopathology-derived patterns extracted from hematoxylin-eosin (H&E) whole-slide images (WSIs) using artificial intelligence (AI) to predict therapeutic response or prognosis. Recently, slide-level multiple-instance learning (MIL) with pathology foundation models (PFMs) has become the standard baseline for CB development. While these methods have improved predictive performance, computational pathology lacks standardized intermediate data formats, provenance tracking, checkpointing conventions, and reproducible evaluation metrics required for clinical-grade deployment. We introduce GOLDMARK (https://artificialintelligencepathology.org), a standardized benchmarking framework built on a curated TCGA cohort with clinically actionable OncoKB level 1-3 biomarker labels. GOLDMARK releases structured intermediate representations, including tile coordinate maps, per-slide feature embeddings from canonical PFMs, quality-control metadata, predefined patient-level splits, trained slide-level models, and evaluation outputs. Models are trained on TCGA and evaluated on an independent MSKCC cohort with reciprocal testing. Across 33 tumor-biomarker tasks, mean AUROC was 0.689 (TCGA) and 0.630 (MSKCC). Restricting to the eight highest-performing tasks yielded mean AUROCs of 0.831 and 0.801, respectively. These tasks correspond to established morphologic-genomic associations (e.g., LGG IDH1, COAD MSI/BRAF, THCA BRAF/NRAS, BLCA FGFR3, UCEC PTEN) and showed the most stable cross-site performance. Differences between canonical encoders were modest relative to task-specific variability. GOLDMARK establishes a shared experimental substrate for computational pathology, enabling reproducible benchmarking and direct comparison of methods across datasets and models.
Single GPU Task Adaptation of Pathology Foundation Models for Whole Slide Image Analysis
Jun 05, 2025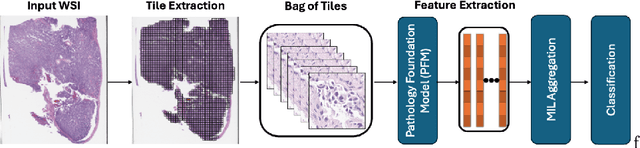
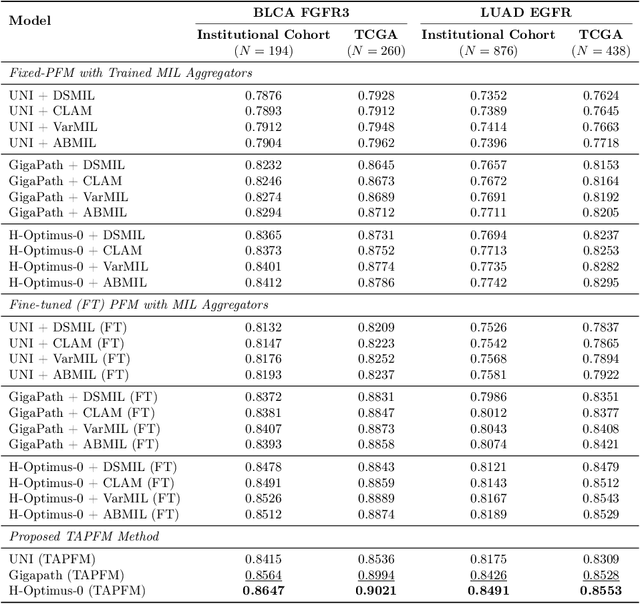
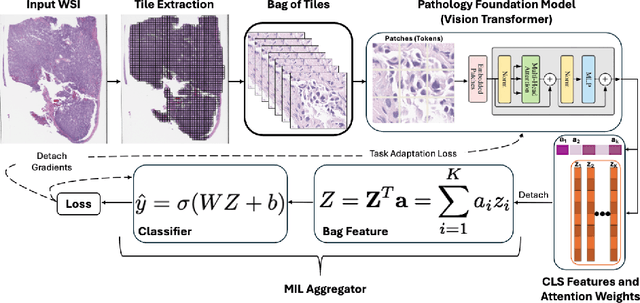
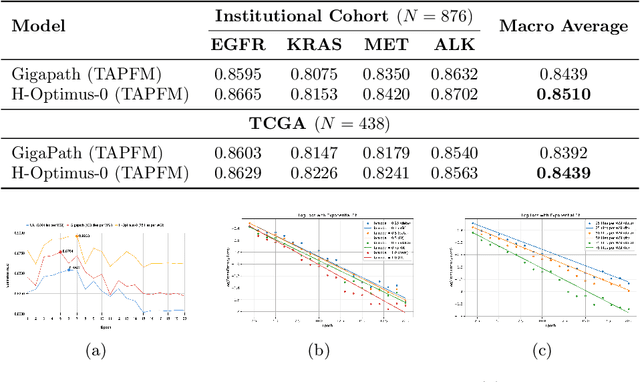
Pathology foundation models (PFMs) have emerged as powerful tools for analyzing whole slide images (WSIs). However, adapting these pretrained PFMs for specific clinical tasks presents considerable challenges, primarily due to the availability of only weak (WSI-level) labels for gigapixel images, necessitating multiple instance learning (MIL) paradigm for effective WSI analysis. This paper proposes a novel approach for single-GPU \textbf{T}ask \textbf{A}daptation of \textbf{PFM}s (TAPFM) that uses vision transformer (\vit) attention for MIL aggregation while optimizing both for feature representations and attention weights. The proposed approach maintains separate computational graphs for MIL aggregator and the PFM to create stable training dynamics that align with downstream task objectives during end-to-end adaptation. Evaluated on mutation prediction tasks for bladder cancer and lung adenocarcinoma across institutional and TCGA cohorts, TAPFM consistently outperforms conventional approaches, with H-Optimus-0 (TAPFM) outperforming the benchmarks. TAPFM effectively handles multi-label classification of actionable mutations as well. Thus, TAPFM makes adaptation of powerful pre-trained PFMs practical on standard hardware for various clinical applications.