 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOne Model is All You Need: Multi-Task Learning Enables Simultaneous Histology Image Segmentation and Classification
Feb 28, 2022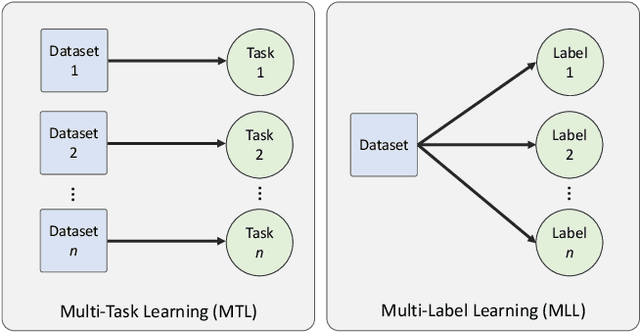
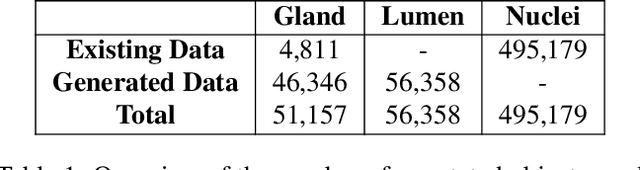
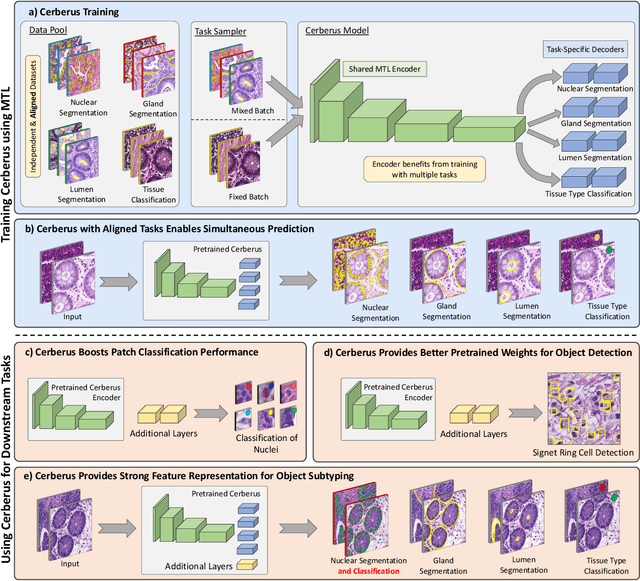
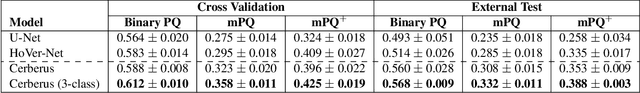
The recent surge in performance for image analysis of digitised pathology slides can largely be attributed to the advance of deep learning. Deep models can be used to initially localise various structures in the tissue and hence facilitate the extraction of interpretable features for biomarker discovery. However, these models are typically trained for a single task and therefore scale poorly as we wish to adapt the model for an increasing number of different tasks. Also, supervised deep learning models are very data hungry and therefore rely on large amounts of training data to perform well. In this paper we present a multi-task learning approach for segmentation and classification of nuclei, glands, lumen and different tissue regions that leverages data from multiple independent data sources. While ensuring that our tasks are aligned by the same tissue type and resolution, we enable simultaneous prediction with a single network. As a result of feature sharing, we also show that the learned representation can be used to improve downstream tasks, including nuclear classification and signet ring cell detection. As part of this work, we use a large dataset consisting of over 600K objects for segmentation and 440K patches for classification and make the data publicly available. We use our approach to process the colorectal subset of TCGA, consisting of 599 whole-slide images, to localise 377 million, 900K and 2.1 million nuclei, glands and lumen respectively. We make this resource available to remove a major barrier in the development of explainable models for computational pathology.
Handcrafted Histological Transformer (H2T): Unsupervised Representation of Whole Slide Images
Feb 14, 2022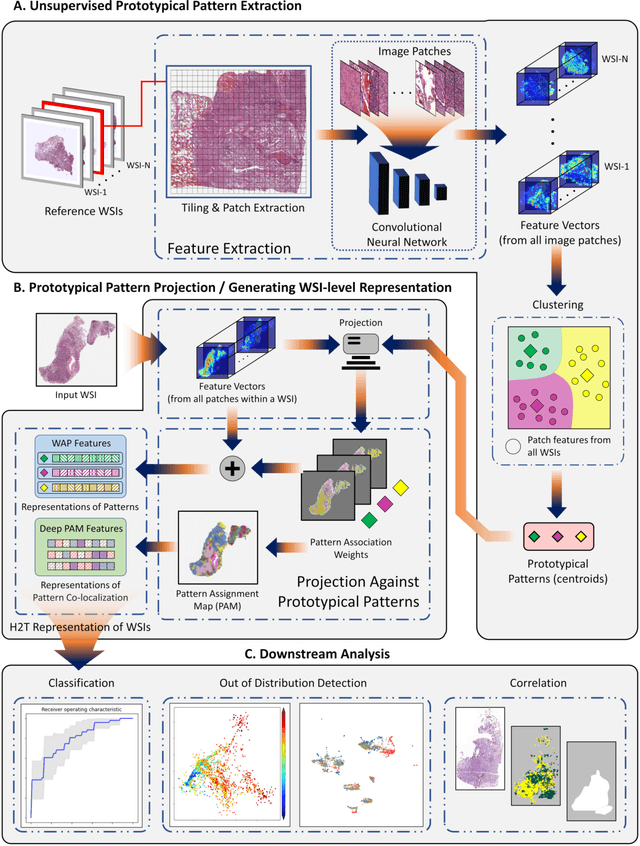
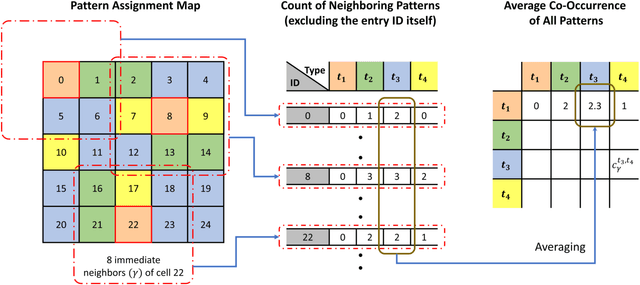
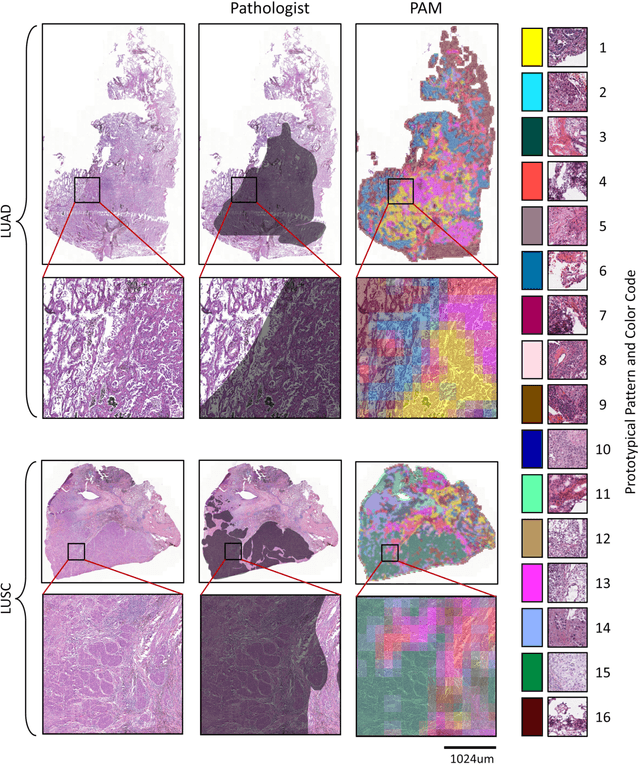
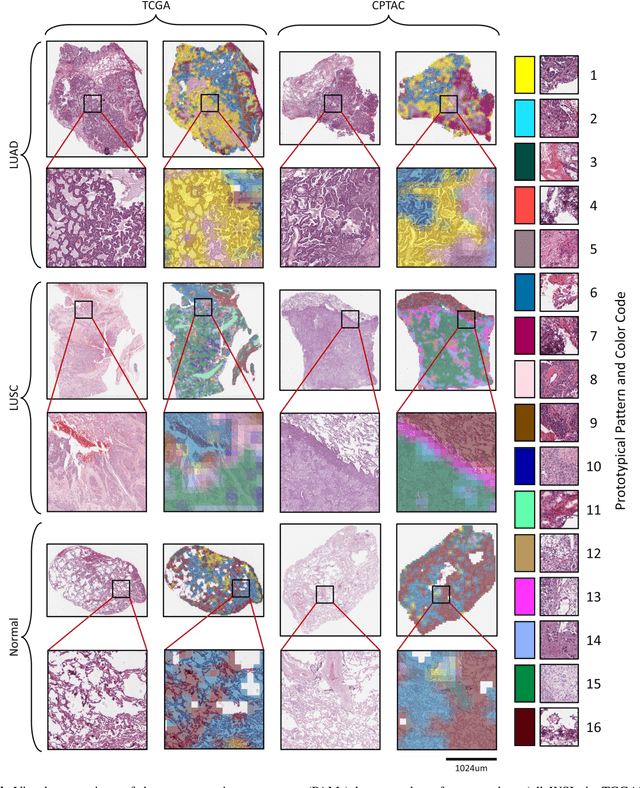
Diagnostic, prognostic and therapeutic decision-making of cancer in pathology clinics can now be carried out based on analysis of multi-gigapixel tissue images, also known as whole-slide images (WSIs). Recently, deep convolutional neural networks (CNNs) have been proposed to derive unsupervised WSI representations; these are attractive as they rely less on expert annotation which is cumbersome. However, a major trade-off is that higher predictive power generally comes at the cost of interpretability, posing a challenge to their clinical use where transparency in decision-making is generally expected. To address this challenge, we present a handcrafted framework based on deep CNN for constructing holistic WSI-level representations. Building on recent findings about the internal working of the Transformer in the domain of natural language processing, we break down its processes and handcraft them into a more transparent framework that we term as the Handcrafted Histological Transformer or H2T. Based on our experiments involving various datasets consisting of a total of 5,306 WSIs, the results demonstrate that H2T based holistic WSI-level representations offer competitive performance compared to recent state-of-the-art methods and can be readily utilized for various downstream analysis tasks. Finally, our results demonstrate that the H2T framework can be up to 14 times faster than the Transformer models.
On Smart Gaze based Annotation of Histopathology Images for Training of Deep Convolutional Neural Networks
Feb 06, 2022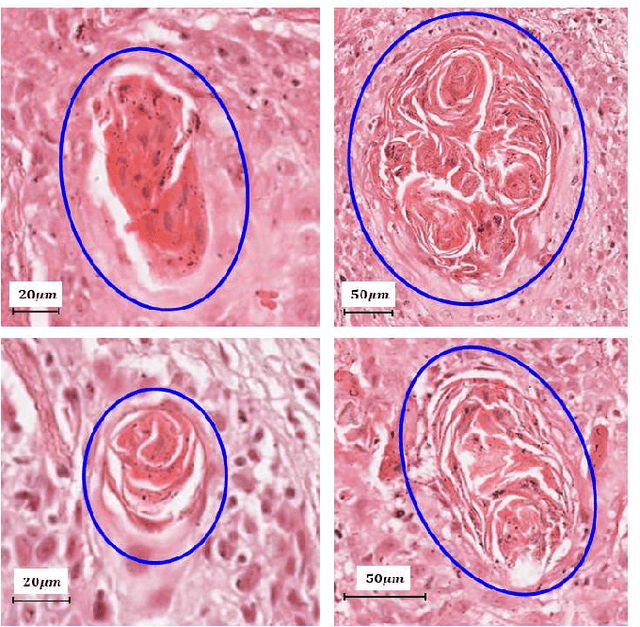
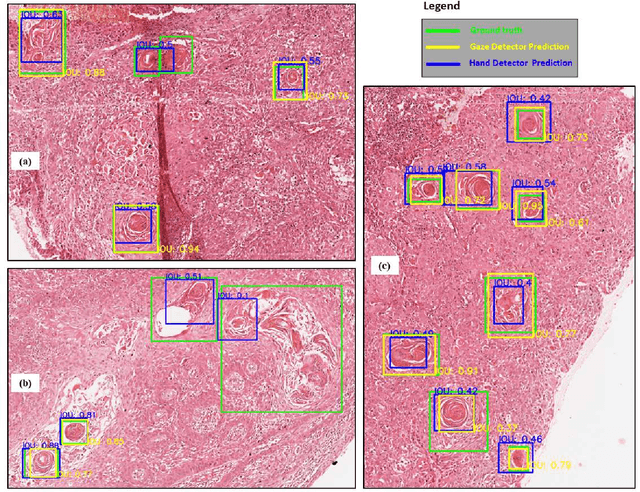
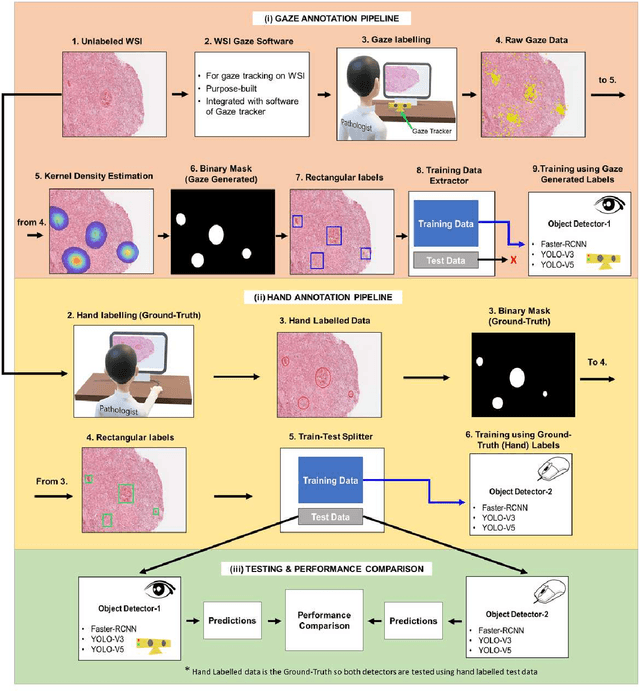
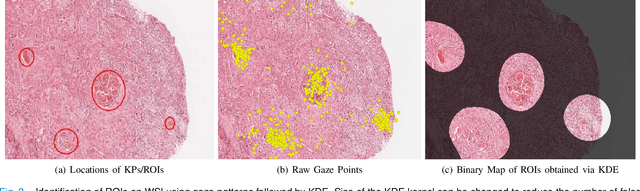
Unavailability of large training datasets is a bottleneck that needs to be overcome to realize the true potential of deep learning in histopathology applications. Although slide digitization via whole slide imaging scanners has increased the speed of data acquisition, labeling of virtual slides requires a substantial time investment from pathologists. Eye gaze annotations have the potential to speed up the slide labeling process. This work explores the viability and timing comparisons of eye gaze labeling compared to conventional manual labeling for training object detectors. Challenges associated with gaze based labeling and methods to refine the coarse data annotations for subsequent object detection are also discussed. Results demonstrate that gaze tracking based labeling can save valuable pathologist time and delivers good performance when employed for training a deep object detector. Using the task of localization of Keratin Pearls in cases of oral squamous cell carcinoma as a test case, we compare the performance gap between deep object detectors trained using hand-labelled and gaze-labelled data. On average, compared to `Bounding-box' based hand-labeling, gaze-labeling required $57.6\%$ less time per label and compared to `Freehand' labeling, gaze-labeling required on average $85\%$ less time per label.
REET: Robustness Evaluation and Enhancement Toolbox for Computational Pathology
Jan 28, 2022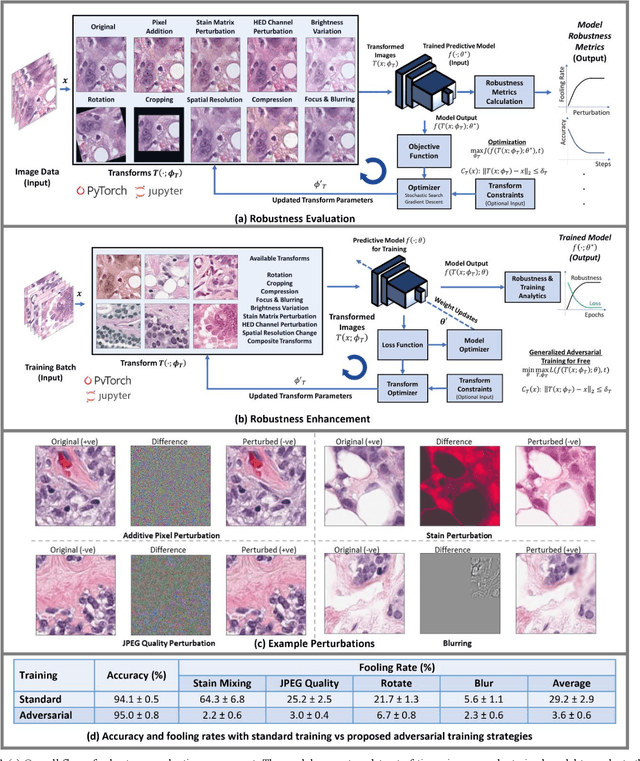
Motivation: Digitization of pathology laboratories through digital slide scanners and advances in deep learning approaches for objective histological assessment have resulted in rapid progress in the field of computational pathology (CPath) with wide-ranging applications in medical and pharmaceutical research as well as clinical workflows. However, the estimation of robustness of CPath models to variations in input images is an open problem with a significant impact on the down-stream practical applicability, deployment and acceptability of these approaches. Furthermore, development of domain-specific strategies for enhancement of robustness of such models is of prime importance as well. Implementation and Availability: In this work, we propose the first domain-specific Robustness Evaluation and Enhancement Toolbox (REET) for computational pathology applications. It provides a suite of algorithmic strategies for enabling robustness assessment of predictive models with respect to specialized image transformations such as staining, compression, focusing, blurring, changes in spatial resolution, brightness variations, geometric changes as well as pixel-level adversarial perturbations. Furthermore, REET also enables efficient and robust training of deep learning pipelines in computational pathology. REET is implemented in Python and is available at the following URL: https://github.com/alexjfoote/reetoolbox. Contact: Fayyaz.minhas@warwick.ac.uk
Towards Launching AI Algorithms for Cellular Pathology into Clinical & Pharmaceutical Orbits
Dec 17, 2021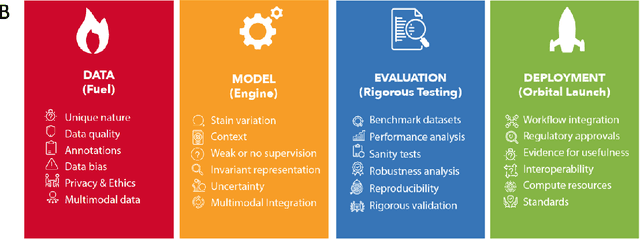

Computational Pathology (CPath) is an emerging field concerned with the study of tissue pathology via computational algorithms for the processing and analysis of digitized high-resolution images of tissue slides. Recent deep learning based developments in CPath have successfully leveraged sheer volume of raw pixel data in histology images for predicting target parameters in the domains of diagnostics, prognostics, treatment sensitivity and patient stratification -- heralding the promise of a new data-driven AI era for both histopathology and oncology. With data serving as the fuel and AI as the engine, CPath algorithms are poised to be ready for takeoff and eventual launch into clinical and pharmaceutical orbits. In this paper, we discuss CPath limitations and associated challenges to enable the readers distinguish hope from hype and provide directions for future research to overcome some of the major challenges faced by this budding field to enable its launch into the two orbits.
CoNIC: Colon Nuclei Identification and Counting Challenge 2022
Nov 29, 2021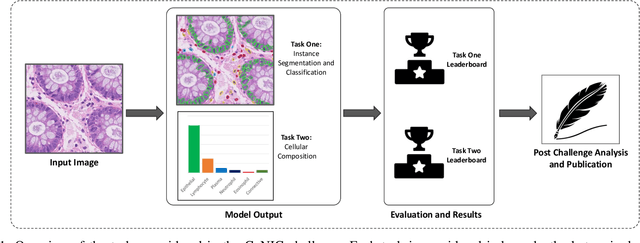
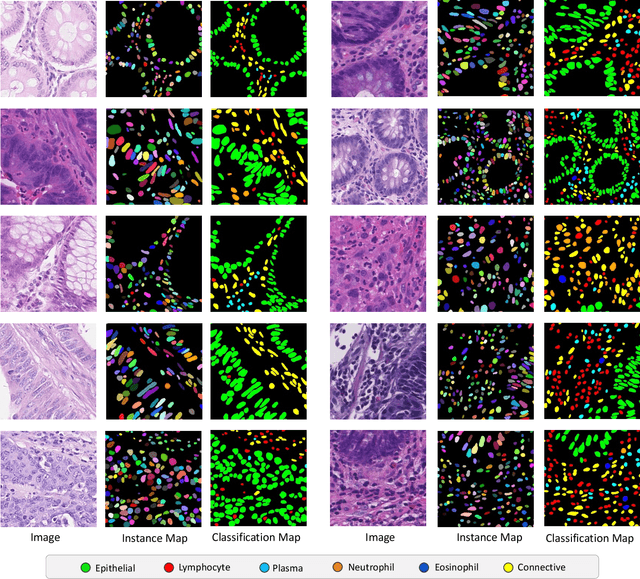
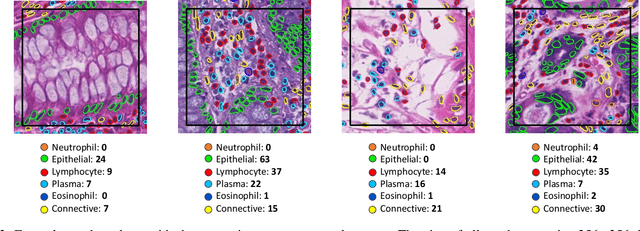
Nuclear segmentation, classification and quantification within Haematoxylin & Eosin stained histology images enables the extraction of interpretable cell-based features that can be used in downstream explainable models in computational pathology (CPath). However, automatic recognition of different nuclei is faced with a major challenge in that there are several different types of nuclei, some of them exhibiting large intra-class variability. To help drive forward research and innovation for automatic nuclei recognition in CPath, we organise the Colon Nuclei Identification and Counting (CoNIC) Challenge. The challenge encourages researchers to develop algorithms that perform segmentation, classification and counting of nuclei within the current largest known publicly available nuclei-level dataset in CPath, containing around half a million labelled nuclei. Therefore, the CoNIC challenge utilises over 10 times the number of nuclei as the previous largest challenge dataset for nuclei recognition. It is important for algorithms to be robust to input variation if we wish to deploy them in a clinical setting. Therefore, as part of this challenge we will also test the sensitivity of each submitted algorithm to certain input variations.
SlideGraph+: Whole Slide Image Level Graphs to Predict HER2Status in Breast Cancer
Oct 12, 2021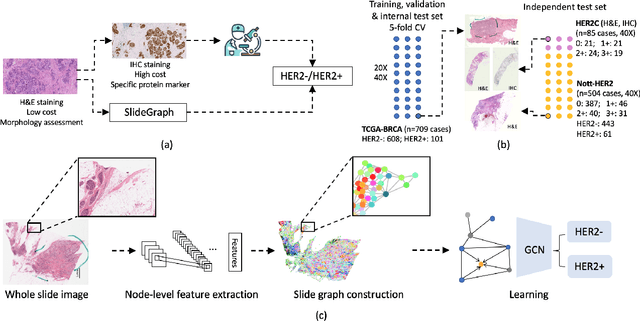
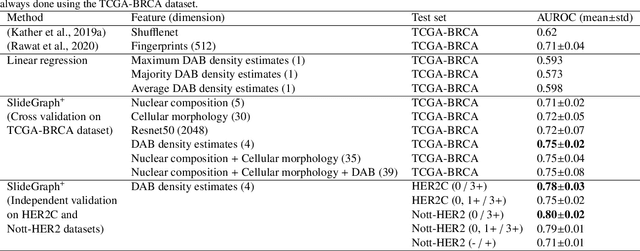
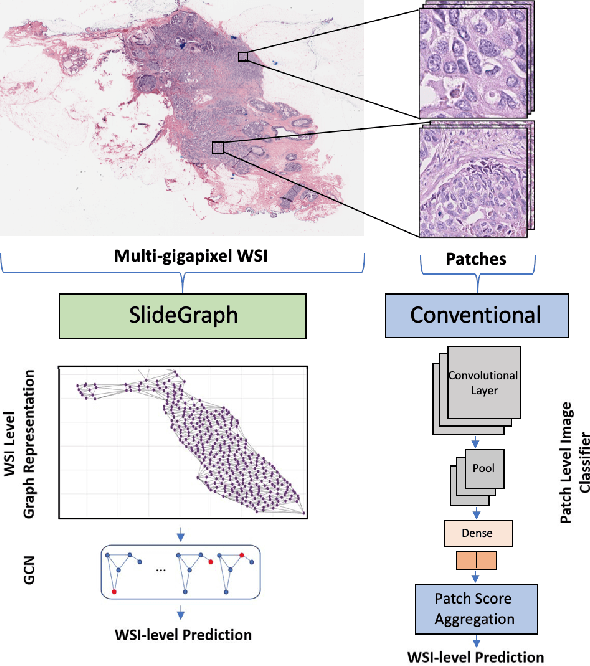

Human epidermal growth factor receptor 2 (HER2) is an important prognostic and predictive factor which is overexpressed in 15-20% of breast cancer (BCa). The determination of its status is a key clinical decision making step for selection of treatment regimen and prognostication. HER2 status is evaluated using transcroptomics or immunohistochemistry (IHC) through situ hybridisation (ISH) which require additional costs and tissue burden in addition to analytical variabilities in terms of manual observational biases in scoring. In this study, we propose a novel graph neural network (GNN) based model (termed SlideGraph+) to predict HER2 status directly from whole-slide images of routine Haematoxylin and Eosin (H&E) slides. The network was trained and tested on slides from The Cancer Genome Atlas (TCGA) in addition to two independent test datasets. We demonstrate that the proposed model outperforms the state-of-the-art methods with area under the ROC curve (AUC) values > 0.75 on TCGA and 0.8 on independent test sets. Our experiments show that the proposed approach can be utilised for case triaging as well as pre-ordering diagnostic tests in a diagnostic setting. It can also be used for other weakly supervised prediction problems in computational pathology. The SlideGraph+ code is available at https://github.com/wenqi006/SlideGraph.
Stain-Robust Mitotic Figure Detection for the Mitosis Domain Generalization Challenge
Sep 29, 2021
The detection of mitotic figures from different scanners/sites remains an important topic of research, owing to its potential in assisting clinicians with tumour grading. The MItosis DOmain Generalization (MIDOG) challenge aims to test the robustness of detection models on unseen data from multiple scanners for this task. We present a short summary of the approach employed by the TIA Centre team to address this challenge. Our approach is based on a hybrid detection model, where mitotic candidates are segmented on stain normalised images, before being refined by a deep learning classifier. Cross-validation on the training images achieved the F1-score of 0.786 and 0.765 on the preliminary test set, demonstrating the generalizability of our model to unseen data from new scanners.
Robust Interactive Semantic Segmentation of Pathology Images with Minimal User Input
Aug 30, 2021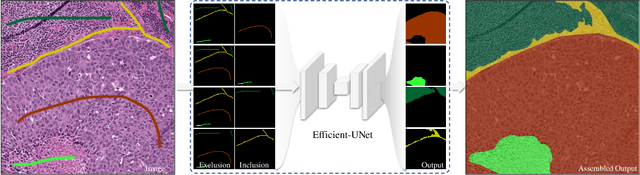

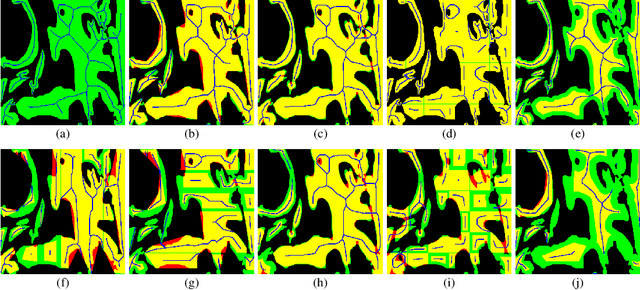
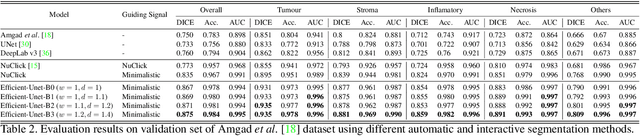
From the simple measurement of tissue attributes in pathology workflow to designing an explainable diagnostic/prognostic AI tool, access to accurate semantic segmentation of tissue regions in histology images is a prerequisite. However, delineating different tissue regions manually is a laborious, time-consuming and costly task that requires expert knowledge. On the other hand, the state-of-the-art automatic deep learning models for semantic segmentation require lots of annotated training data and there are only a limited number of tissue region annotated images publicly available. To obviate this issue in computational pathology projects and collect large-scale region annotations efficiently, we propose an efficient interactive segmentation network that requires minimum input from the user to accurately annotate different tissue types in the histology image. The user is only required to draw a simple squiggle inside each region of interest so it will be used as the guiding signal for the model. To deal with the complex appearance and amorph geometry of different tissue regions we introduce several automatic and minimalistic guiding signal generation techniques that help the model to become robust against the variation in the user input. By experimenting on a dataset of breast cancer images, we show that not only does our proposed method speed up the interactive annotation process, it can also outperform the existing automatic and interactive region segmentation models.
Lizard: A Large-Scale Dataset for Colonic Nuclear Instance Segmentation and Classification
Aug 25, 2021
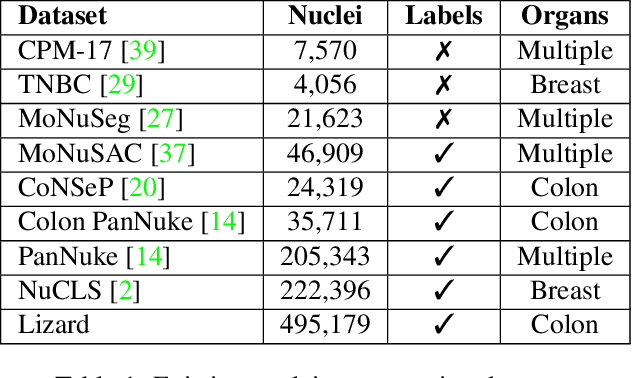
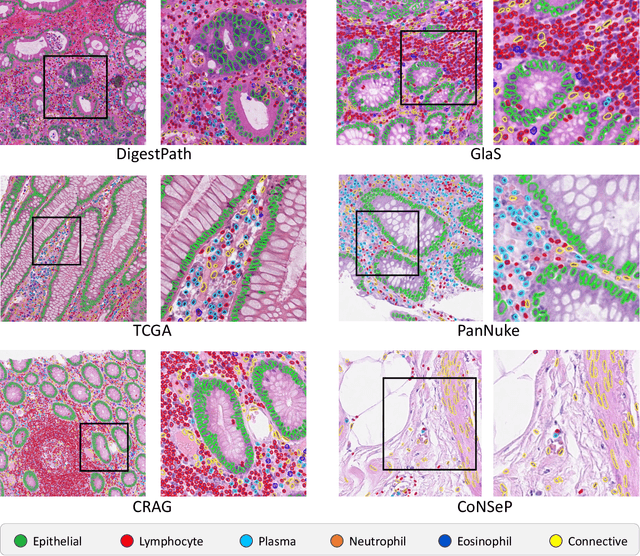
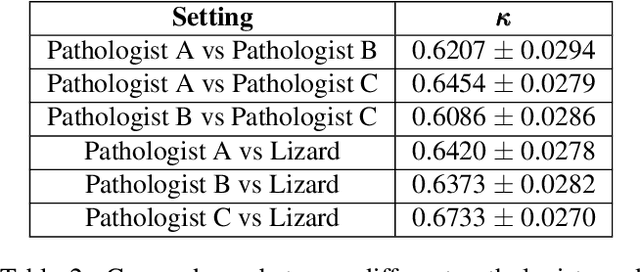
The development of deep segmentation models for computational pathology (CPath) can help foster the investigation of interpretable morphological biomarkers. Yet, there is a major bottleneck in the success of such approaches because supervised deep learning models require an abundance of accurately labelled data. This issue is exacerbated in the field of CPath because the generation of detailed annotations usually demands the input of a pathologist to be able to distinguish between different tissue constructs and nuclei. Manually labelling nuclei may not be a feasible approach for collecting large-scale annotated datasets, especially when a single image region can contain thousands of different cells. However, solely relying on automatic generation of annotations will limit the accuracy and reliability of ground truth. Therefore, to help overcome the above challenges, we propose a multi-stage annotation pipeline to enable the collection of large-scale datasets for histology image analysis, with pathologist-in-the-loop refinement steps. Using this pipeline, we generate the largest known nuclear instance segmentation and classification dataset, containing nearly half a million labelled nuclei in H&E stained colon tissue. We have released the dataset and encourage the research community to utilise it to drive forward the development of downstream cell-based models in CPath.