 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFrom Words to Amino Acids: Does the Curse of Depth Persist?
Feb 25, 2026Protein language models (PLMs) have become widely adopted as general-purpose models, demonstrating strong performance in protein engineering and de novo design. Like large language models (LLMs), they are typically trained as deep transformers with next-token or masked-token prediction objectives on massive sequence corpora and are scaled by increasing model depth. Recent work on autoregressive LLMs has identified the Curse of Depth: later layers contribute little to the final output predictions. These findings naturally raise the question of whether a similar depth inefficiency also appears in PLMs, where many widely used models are not autoregressive, and some are multimodal, accepting both protein sequence and structure as input. In this work, we present a depth analysis of six popular PLMs across model families and scales, spanning three training objectives, namely autoregressive, masked, and diffusion, and quantify how layer contributions evolve with depth using a unified set of probing- and perturbation-based measurements. Across all models, we observe consistent depth-dependent patterns that extend prior findings on LLMs: later layers depend less on earlier computations and mainly refine the final output distribution, and these effects are increasingly pronounced in deeper models. Taken together, our results suggest that PLMs exhibit a form of depth inefficiency, motivating future work on more depth-efficient architectures and training methods.
ProtTrans: Towards Cracking the Language of Life's Code Through Self-Supervised Deep Learning and High Performance Computing
Jul 20, 2020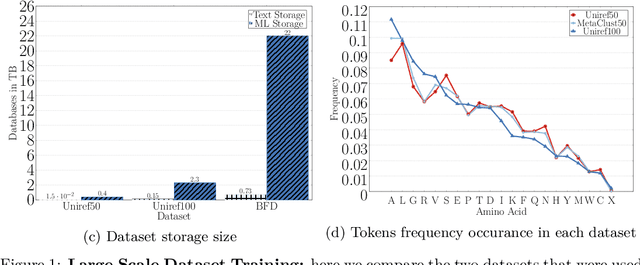
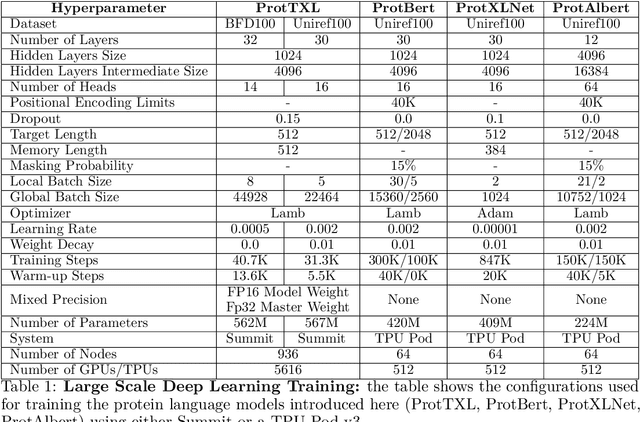
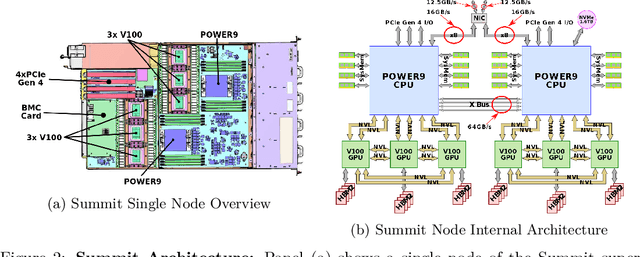
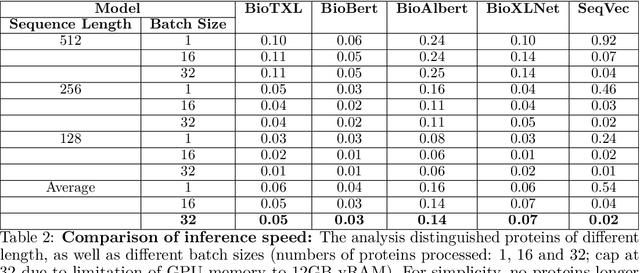
Computational biology and bioinformatics provide vast data gold-mines from protein sequences, ideal for Language Models (LMs) taken from Natural Language Processing (NLP). These LMs reach for new prediction frontiers at low inference costs. Here, we trained two auto-regressive language models (Transformer-XL, XLNet) and two auto-encoder models (Bert, Albert) on data from UniRef and BFD containing up to 393 billion amino acids (words) from 2.1 billion protein sequences (22- and 112-times the entire English Wikipedia). The LMs were trained on the Summit supercomputer at Oak Ridge National Laboratory (ORNL), using 936 nodes (total 5616 GPUs) and one TPU Pod (V3-512 or V3-1024). We validated the advantage of up-scaling LMs to larger models supported by bigger data by predicting secondary structure (3-states: Q3=76-84, 8-states: Q8=65-73), sub-cellular localization for 10 cellular compartments (Q10=74) and whether a protein is membrane-bound or water-soluble (Q2=89). Dimensionality reduction revealed that the LM-embeddings from unlabeled data (only protein sequences) captured important biophysical properties governing protein shape. This implied learning some of the grammar of the language of life realized in protein sequences. The successful up-scaling of protein LMs through HPC to larger data sets slightly reduced the gap between models trained on evolutionary information and LMs. The official GitHub repository: https://github.com/agemagician/ProtTrans