 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSLSREC: Self-Supervised Contrastive Learning for Adaptive Fusion of Long- and Short-Term User Interests
Apr 06, 2026User interests typically encompass both long-term preferences and short-term intentions, reflecting the dynamic nature of user behaviors across different timeframes. The uneven temporal distribution of user interactions highlights the evolving patterns of interests, making it challenging to accurately capture shifts in interests using comprehensive historical behaviors. To address this, we propose SLSRec, a novel Session-based model with the fusion of Long- and Short-term Recommendations that effectively captures the temporal dynamics of user interests by segmenting historical behaviors over time. Unlike conventional models that combine long- and short-term user interests into a single representation, compromising recommendation accuracy, SLSRec utilizes a self-supervised learning framework to disentangle these two types of interests. A contrastive learning strategy is introduced to ensure accurate calibration of long- and short-term interest representations. Additionally, an attention-based fusion network is designed to adaptively aggregate interest representations, optimizing their integration to enhance recommendation performance. Extensive experiments on three public benchmark datasets demonstrate that SLSRec consistently outperforms state-of-the-art models while exhibiting superior robustness across various scenarios.We will release all source code upon acceptance.
Learning Task-Invariant Properties via Dreamer: Enabling Efficient Policy Transfer for Quadruped Robots
Apr 03, 2026Achieving quadruped robot locomotion across diverse and dynamic terrains presents significant challenges, primarily due to the discrepancies between simulation environments and real-world conditions. Traditional sim-to-real transfer methods often rely on manual feature design or costly real-world fine-tuning. To address these limitations, this paper proposes the DreamTIP framework, which incorporates Task-Invariant Properties learning within the Dreamer world model architecture to enhance sim-to-real transfer capabilities. Guided by large language models, DreamTIP identifies and leverages Task-Invariant Properties, such as contact stability and terrain clearance, which exhibit robustness to dynamic variations and strong transferability across tasks. These properties are integrated into the world model as auxiliary prediction targets, enabling the policy to learn representations that are insensitive to underlying dynamic changes. Furthermore, an efficient adaptation strategy is designed, employing a mixed replay buffer and regularization constraints to rapidly calibrate to real-world dynamics while effectively mitigating representation collapse and catastrophic forgetting. Extensive experiments on complex terrains, including Stair, Climb, Tilt, and Crawl, demonstrate that DreamTIP significantly outperforms state-of-the-art baselines in both simulated and real-world environments. Our method achieves an average performance improvement of 28.1% across eight distinct simulated transfer tasks. In the real-world Climb task, the baseline method achieved only a 10\ success rate, whereas our method attained a 100% success rate. These results indicate that incorporating Task-Invariant Properties into Dreamer learning offers a novel solution for achieving robust and transferable robot locomotion.
PreferRec: Learning and Transferring Pareto Preferences for Multi-objective Re-ranking
Mar 23, 2026Multi-objective re-ranking has become a critical component of modern multi-stage recommender systems, as it tasked to balance multiple conflicting objectives such as accuracy, diversity, and fairness. Existing multi-objective re-ranking methods typically optimize aggregate objectives at the item level using static or handcrafted preference weights. This design overlooks that users inherently exhibit Pareto-optimal preferences at the intent level, reflecting personalized trade-offs among objectives rather than fixed weight combinations. Moreover, most approaches treat re-ranking task for each user as an isolated problem, and repeatedly learn the preferences from scratch. Such a paradigm not only incurs high computational cost, but also ignores the fact that users often share similar preference trade-off structures across objectives. Inspired by the existence of homogeneous multi-objective optimization spaces where Pareto-optimal patterns are transferable, we propose PreferRec, a novel framework that explicitly models and transfers Pareto preferences across users. Specifically, PreferRec is built upon three tightly coupled components: Preference-Aware Pareto Learning aims to capture user intrinsic trade-offs among multiple conflicting objectives at the intent level. By learning Pareto preference representations from re-ranking populations, this component explicitly models how users prioritize different objectives under diverse contexts. Knowledge-Guided Transfer facilitates efficient cross-user knowledge transfer by distilling shared optimization patterns across homogeneous optimization spaces. The transferred knowledge is then used to guide solution selection and personalized re-ranking, biasing the optimization process toward high-quality regions of the Pareto front while preserving user-specific preference characteristics.
RIGA-Fold: A General Framework for Protein Inverse Folding via Recurrent Interaction and Geometric Awareness
Feb 04, 2026Protein inverse folding, the task of predicting amino acid sequences for desired structures, is pivotal for de novo protein design. However, existing GNN-based methods typically suffer from restricted receptive fields that miss long-range dependencies and a "single-pass" inference paradigm that leads to error accumulation. To address these bottlenecks, we propose RIGA-Fold, a framework that synergizes Recurrent Interaction with Geometric Awareness. At the micro-level, we introduce a Geometric Attention Update (GAU) module where edge features explicitly serve as attention keys, ensuring strictly SE(3)-invariant local encoding. At the macro-level, we design an attention-based Global Context Bridge that acts as a soft gating mechanism to dynamically inject global topological information. Furthermore, to bridge the gap between structural and sequence modalities, we introduce an enhanced variant, RIGA-Fold*, which integrates trainable geometric features with frozen evolutionary priors from ESM-2 and ESM-IF via a dual-stream architecture. Finally, a biologically inspired ``predict-recycle-refine'' strategy is implemented to iteratively denoise sequence distributions. Extensive experiments on CATH 4.2, TS50, and TS500 benchmarks demonstrate that our geometric framework is highly competitive, while RIGA-Fold* significantly outperforms state-of-the-art baselines in both sequence recovery and structural consistency.
Unveiling Scaling Behaviors in Molecular Language Models: Effects of Model Size, Data, and Representation
Jan 30, 2026Molecular generative models, often employing GPT-style language modeling on molecular string representations, have shown promising capabilities when scaled to large datasets and model sizes. However, it remains unclear and subject to debate whether these models adhere to predictable scaling laws under fixed computational budgets, which is a crucial understanding for optimally allocating resources between model size, data volume, and molecular representation. In this study, we systematically investigate the scaling behavior of molecular language models across both pretraining and downstream tasks. We train 300 models and conduct over 10,000 experiments, rigorously controlling compute budgets while independently varying model size, number of training tokens, and molecular representation. Our results demonstrate clear scaling laws in molecular models for both pretraining and downstream transfer, reveal the substantial impact of molecular representation on performance, and explain previously observed inconsistencies in scaling behavior for molecular generation. Additionally, we publicly release the largest library of molecular language models to date to facilitate future research and development. Code and models are available at https://github.com/SZU-ADDG/MLM-Scaling.
From Tokens to Blocks: A Block-Diffusion Perspective on Molecular Generation
Jan 29, 2026Drug discovery can be viewed as a combinatorial search over an immense chemical space, motivating the development of deep generative models for de novo molecular design. Among these, GPT-based molecular language models (MLM) have shown strong molecular design performance by learning chemical syntax and semantics from large-scale data. However, existing MLMs face two fundamental limitations: they inadequately capture the graph-structured nature of molecules when formulated as next-token prediction problems, and they typically lack explicit mechanisms for target-aware generation. Here, we propose SoftMol, a unified framework that co-designs molecular representation, model architecture, and search strategy for target-aware molecular generation. SoftMol introduces soft fragments, a rule-free block representation of SMILES that enables diffusion-native modeling, and develops SoftBD, the first block-diffusion molecular language model that combines local bidirectional diffusion with autoregressive generation under molecular structural constraints. To favor generated molecules with high drug-likeness and synthetic accessibility, SoftBD is trained on a carefully curated dataset named ZINC-Curated. SoftMol further integrates a gated Monte Carlo tree search to assemble fragments in a target-aware manner. Experimental results show that, compared with current state-of-the-art models, SoftMol achieves 100% chemical validity, improves binding affinity by 9.7%, yields a 2-3x increase in molecular diversity, and delivers a 6.6x speedup in inference efficiency. Code is available at https://github.com/szu-aicourse/softmol
Rethinking Drug-Drug Interaction Modeling as Generalizable Relation Learning
Jan 22, 2026Drug-drug interaction (DDI) prediction is central to drug discovery and clinical development, particularly in the context of increasingly prevalent polypharmacy. Although existing computational methods achieve strong performance on standard benchmarks, they often fail to generalize to realistic deployment scenarios, where most candidate drug pairs involve previously unseen drugs and validated interactions are scarce. We demonstrate that proximity in the embedding spaces of prevailing molecule-centric DDI models does not reliably correspond to interaction labels, and that simply scaling up model capacity therefore fails to improve generalization. To address these limitations, we propose GenRel-DDI, a generalizable relation learning framework that reformulates DDI prediction as a relation-centric learning problem, in which interaction representations are learned independently of drug identities. This relation-level abstraction enables the capture of transferable interaction patterns that generalize to unseen drugs and novel drug pairs. Extensive experiments across multiple benchmark demonstrate that GenRel-DDI consistently and significantly outperforms state-of-the-art methods, with particularly large gains on strict entity-disjoint evaluations, highlighting the effectiveness and practical utility of relation learning for robust DDI prediction. The code is available at https://github.com/SZU-ADDG/GenRel-DDI.
Toward Closed-loop Molecular Discovery via Language Model, Property Alignment and Strategic Search
Dec 18, 2025Drug discovery is a time-consuming and expensive process, with traditional high-throughput and docking-based virtual screening hampered by low success rates and limited scalability. Recent advances in generative modelling, including autoregressive, diffusion, and flow-based approaches, have enabled de novo ligand design beyond the limits of enumerative screening. Yet these models often suffer from inadequate generalization, limited interpretability, and an overemphasis on binding affinity at the expense of key pharmacological properties, thereby restricting their translational utility. Here we present Trio, a molecular generation framework integrating fragment-based molecular language modeling, reinforcement learning, and Monte Carlo tree search, for effective and interpretable closed-loop targeted molecular design. Through the three key components, Trio enables context-aware fragment assembly, enforces physicochemical and synthetic feasibility, and guides a balanced search between the exploration of novel chemotypes and the exploitation of promising intermediates within protein binding pockets. Experimental results show that Trio reliably achieves chemically valid and pharmacologically enhanced ligands, outperforming state-of-the-art approaches with improved binding affinity (+7.85%), drug-likeness (+11.10%) and synthetic accessibility (+12.05%), while expanding molecular diversity more than fourfold. By combining generalization, plausibility, and interpretability, Trio establishes a closed-loop generative paradigm that redefines how chemical space can be navigated, offering a transformative foundation for the next era of AI-driven drug discovery.
MODA: A Unified 3D Diffusion Framework for Multi-Task Target-Aware Molecular Generation
Jul 09, 2025Three-dimensional molecular generators based on diffusion models can now reach near-crystallographic accuracy, yet they remain fragmented across tasks. SMILES-only inputs, two-stage pretrain-finetune pipelines, and one-task-one-model practices hinder stereochemical fidelity, task alignment, and zero-shot transfer. We introduce MODA, a diffusion framework that unifies fragment growing, linker design, scaffold hopping, and side-chain decoration with a Bayesian mask scheduler. During training, a contiguous spatial fragment is masked and then denoised in one pass, enabling the model to learn shared geometric and chemical priors across tasks. Multi-task training yields a universal backbone that surpasses six diffusion baselines and three training paradigms on substructure, chemical property, interaction, and geometry. Model-C reduces ligand-protein clashes and substructure divergences while maintaining Lipinski compliance, whereas Model-B preserves similarity but trails in novelty and binding affinity. Zero-shot de novo design and lead-optimisation tests confirm stable negative Vina scores and high improvement rates without force-field refinement. These results demonstrate that a single-stage multi-task diffusion routine can replace two-stage workflows for structure-based molecular design.
Reimagining Target-Aware Molecular Generation through Retrieval-Enhanced Aligned Diffusion
Jun 17, 2025
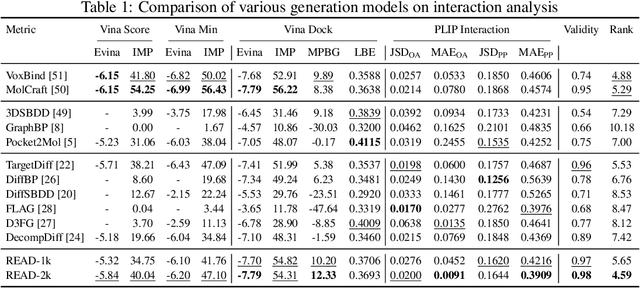
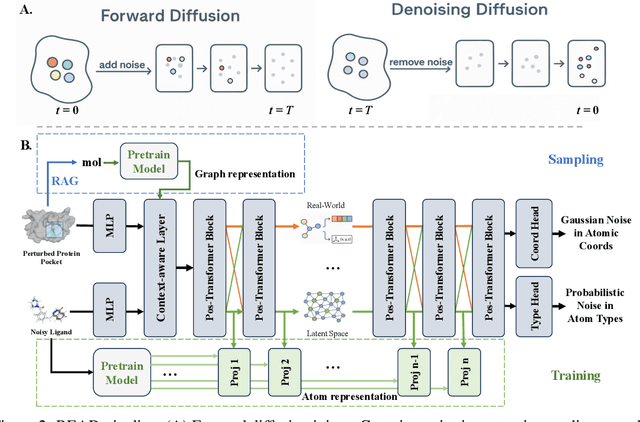
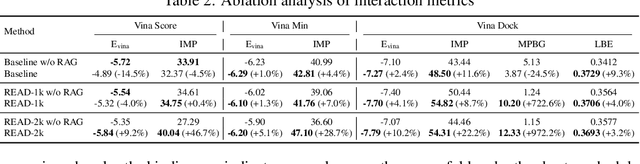
Breakthroughs in high-accuracy protein structure prediction, such as AlphaFold, have established receptor-based molecule design as a critical driver for rapid early-phase drug discovery. However, most approaches still struggle to balance pocket-specific geometric fit with strict valence and synthetic constraints. To resolve this trade-off, a Retrieval-Enhanced Aligned Diffusion termed READ is introduced, which is the first to merge molecular Retrieval-Augmented Generation with an SE(3)-equivariant diffusion model. Specifically, a contrastively pre-trained encoder aligns atom-level representations during training, then retrieves graph embeddings of pocket-matched scaffolds to guide each reverse-diffusion step at inference. This single mechanism can inject real-world chemical priors exactly where needed, producing valid, diverse, and shape-complementary ligands. Experimental results demonstrate that READ can achieve very competitive performance in CBGBench, surpassing state-of-the-art generative models and even native ligands. That suggests retrieval and diffusion can be co-optimized for faster, more reliable structure-based drug design.