 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Matching: Relax at Your Own Risk
Jan 10, 2015
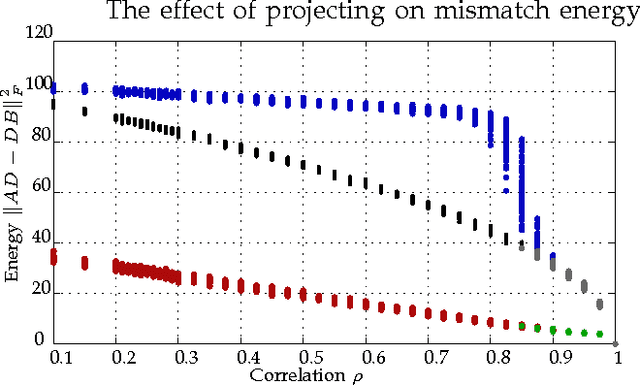
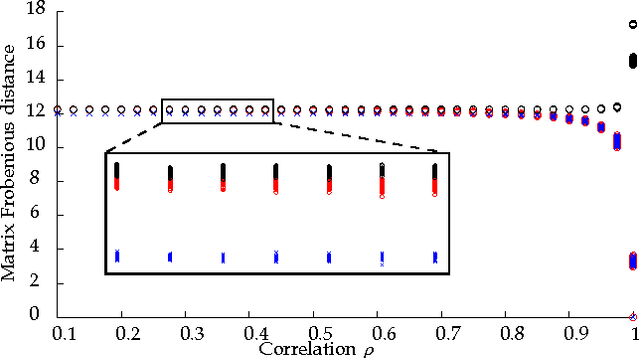
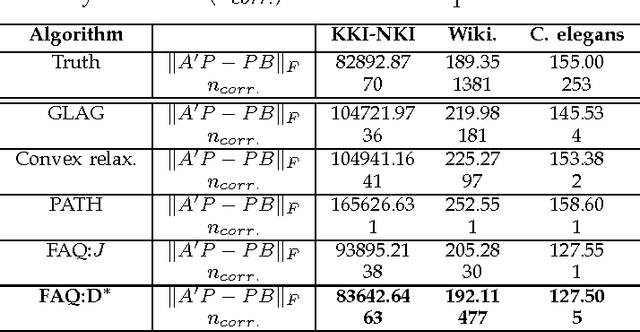
Graph matching---aligning a pair of graphs to minimize their edge disagreements---has received wide-spread attention from both theoretical and applied communities over the past several decades, including combinatorics, computer vision, and connectomics. Its attention can be partially attributed to its computational difficulty. Although many heuristics have previously been proposed in the literature to approximately solve graph matching, very few have any theoretical support for their performance. A common technique is to relax the discrete problem to a continuous problem, therefore enabling practitioners to bring gradient-descent-type algorithms to bear. We prove that an indefinite relaxation (when solved exactly) almost always discovers the optimal permutation, while a common convex relaxation almost always fails to discover the optimal permutation. These theoretical results suggest that initializing the indefinite algorithm with the convex optimum might yield improved practical performance. Indeed, experimental results illuminate and corroborate these theoretical findings, demonstrating that excellent results are achieved in both benchmark and real data problems by amalgamating the two approaches.
Statistical inference on errorfully observed graphs
Jul 21, 2014Statistical inference on graphs is a burgeoning field in the applied and theoretical statistics communities, as well as throughout the wider world of science, engineering, business, etc. In many applications, we are faced with the reality of errorfully observed graphs. That is, the existence of an edge between two vertices is based on some imperfect assessment. In this paper, we consider a graph $G = (V,E)$. We wish to perform an inference task -- the inference task considered here is "vertex classification". However, we do not observe $G$; rather, for each potential edge $uv \in {{V}\choose{2}}$ we observe an "edge-feature" which we use to classify $uv$ as edge/not-edge. Thus we errorfully observe $G$ when we observe the graph $\widetilde{G} = (V,\widetilde{E})$ as the edges in $\widetilde{E}$ arise from the classifications of the "edge-features", and are expected to be errorful. Moreover, we face a quantity/quality trade-off regarding the edge-features we observe -- more informative edge-features are more expensive, and hence the number of potential edges that can be assessed decreases with the quality of the edge-features. We studied this problem by formulating a quantity/quality tradeoff for a simple class of random graphs model, namely the stochastic blockmodel. We then consider a simple but optimal vertex classifier for classifying $v$ and we derive the optimal quantity/quality operating point for subsequent graph inference in the face of this trade-off. The optimal operating points for the quantity/quality trade-off are surprising and illustrate the issue that methods for intermediate tasks should be chosen to maximize performance for the ultimate inference task. Finally, we investigate the quantity/quality tradeoff for errorful obesrvations of the {\it C.\ elegans} connectome graph.
Automatic Annotation of Axoplasmic Reticula in Pursuit of Connectomes
Apr 16, 2014

In this paper, we present a new pipeline which automatically identifies and annotates axoplasmic reticula, which are small subcellular structures present only in axons. We run our algorithm on the Kasthuri11 dataset, which was color corrected using gradient-domain techniques to adjust contrast. We use a bilateral filter to smooth out the noise in this data while preserving edges, which highlights axoplasmic reticula. These axoplasmic reticula are then annotated using a morphological region growing algorithm. Additionally, we perform Laplacian sharpening on the bilaterally filtered data to enhance edges, and repeat the morphological region growing algorithm to annotate more axoplasmic reticula. We track our annotations through the slices to improve precision, and to create long objects to aid in segment merging. This method annotates axoplasmic reticula with high precision. Our algorithm can easily be adapted to annotate axoplasmic reticula in different sets of brain data by changing a few thresholds. The contribution of this work is the introduction of a straightforward and robust pipeline which annotates axoplasmic reticula with high precision, contributing towards advancements in automatic feature annotations in neural EM data.
Seeded Graph Matching Via Joint Optimization of Fidelity and Commensurability
Jan 16, 2014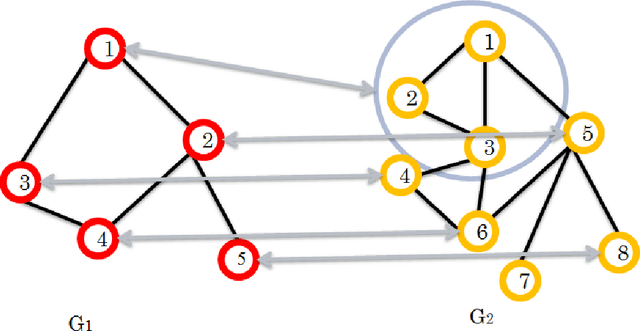
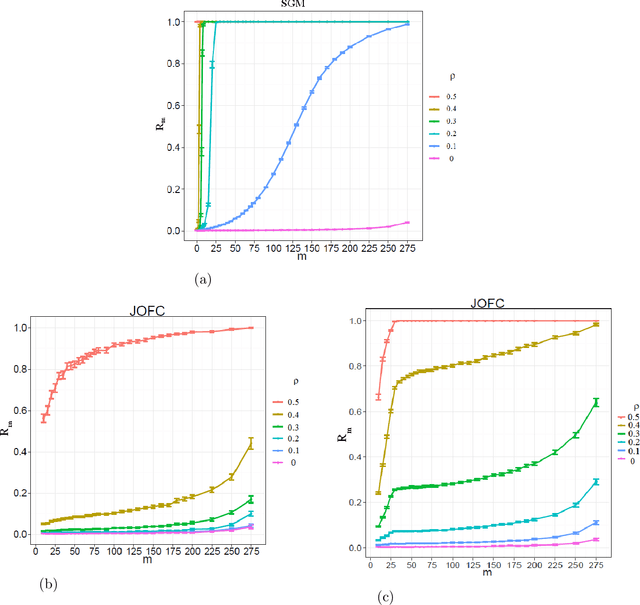
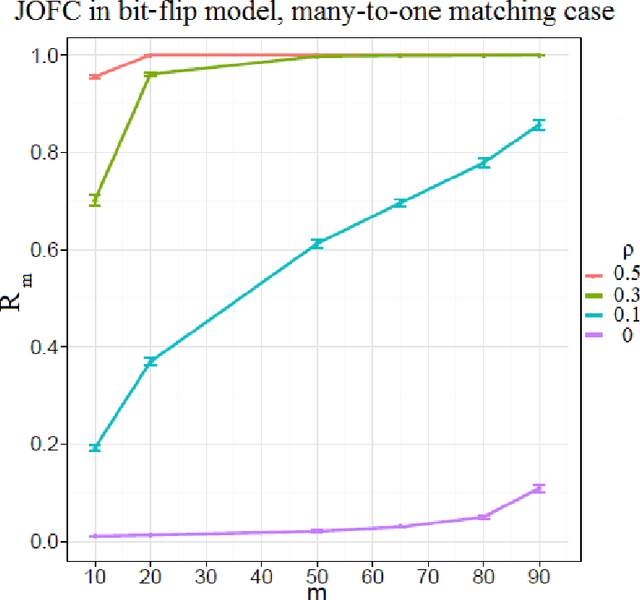
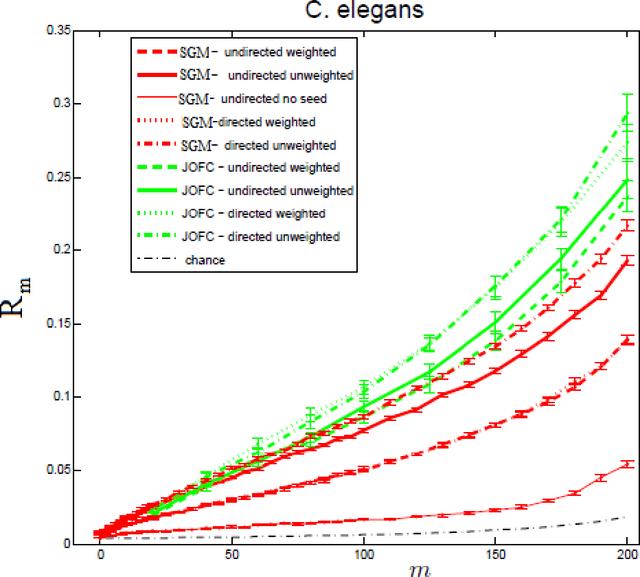
We present a novel approximate graph matching algorithm that incorporates seeded data into the graph matching paradigm. Our Joint Optimization of Fidelity and Commensurability (JOFC) algorithm embeds two graphs into a common Euclidean space where the matching inference task can be performed. Through real and simulated data examples, we demonstrate the versatility of our algorithm in matching graphs with various characteristics--weightedness, directedness, loopiness, many-to-one and many-to-many matchings, and soft seedings.
Bayesian crack detection in ultra high resolution multimodal images of paintings
Apr 23, 2013



The preservation of our cultural heritage is of paramount importance. Thanks to recent developments in digital acquisition techniques, powerful image analysis algorithms are developed which can be useful non-invasive tools to assist in the restoration and preservation of art. In this paper we propose a semi-supervised crack detection method that can be used for high-dimensional acquisitions of paintings coming from different modalities. Our dataset consists of a recently acquired collection of images of the Ghent Altarpiece (1432), one of Northern Europe's most important art masterpieces. Our goal is to build a classifier that is able to discern crack pixels from the background consisting of non-crack pixels, making optimal use of the information that is provided by each modality. To accomplish this we employ a recently developed non-parametric Bayesian classifier, that uses tensor factorizations to characterize any conditional probability. A prior is placed on the parameters of the factorization such that every possible interaction between predictors is allowed while still identifying a sparse subset among these predictors. The proposed Bayesian classifier, which we will refer to as conditional Bayesian tensor factorization or CBTF, is assessed by visually comparing classification results with the Random Forest (RF) algorithm.