 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBioMiner: A Multi-modal System for Automated Mining of Protein-Ligand Bioactivity Data from Literature
Apr 23, 2026Protein-ligand bioactivity data published in the literature are essential for drug discovery, yet manual curation struggles to keep pace with rapidly growing literature. Automated bioactivity extraction remains challenging because it requires not only interpreting biochemical semantics distributed across text, tables, and figures, but also reconstructing chemically exact ligand structures (e.g., Markush structures). To address this bottleneck, we introduce BioMiner, a multi-modal extraction framework that explicitly separates bioactivity semantic interpretation from ligand structure construction. Within BioMiner, bioactivity semantics are inferred through direct reasoning, while chemical structures are resolved via a chemical-structure-grounded visual semantic reasoning paradigm, in which multi-modal large language models operate on chemically grounded visual representations to infer inter-structure relationships, and exact molecular construction is delegated to domain chemistry tools. For rigorous evaluation and method development, we further establish BioVista, a comprehensive benchmark comprising 16,457 bioactivity entries curated from 500 publications. BioMiner validates its extraction ability and provides a quantitative baseline, achieving an F1 score of 0.32 for bioactivity triplets. BioMiner's practical utility is demonstrated via three applications: (1) extracting 82,262 data from 11,683 papers to build a pre-training database that improves downstream models performance by 3.9%; (2) enabling a human-in-the-loop workflow that doubles the number of high-quality NLRP3 bioactivity data, helping 38.6% improvement over 28 QSAR models and identification of 16 hit candidates with novel scaffolds; and (3) accelerating protein-ligand complex bioactivity annotation, achieving a 5.59-fold speed increase and 5.75% accuracy improvement over manual workflows in PoseBusters dataset.
DeltaDock: A Unified Framework for Accurate, Efficient, and Physically Reliable Molecular Docking
Oct 15, 2024Molecular docking, a technique for predicting ligand binding poses, is crucial in structure-based drug design for understanding protein-ligand interactions. Recent advancements in docking methods, particularly those leveraging geometric deep learning (GDL), have demonstrated significant efficiency and accuracy advantages over traditional sampling methods. Despite these advancements, current methods are often tailored for specific docking settings, and limitations such as the neglect of protein side-chain structures, difficulties in handling large binding pockets, and challenges in predicting physically valid structures exist. To accommodate various docking settings and achieve accurate, efficient, and physically reliable docking, we propose a novel two-stage docking framework, DeltaDock, consisting of pocket prediction and site-specific docking. We innovatively reframe the pocket prediction task as a pocket-ligand alignment problem rather than direct prediction in the first stage. Then we follow a bi-level coarse-to-fine iterative refinement process to perform site-specific docking. Comprehensive experiments demonstrate the superior performance of DeltaDock. Notably, in the blind docking setting, DeltaDock achieves a 31\% relative improvement over the docking success rate compared with the previous state-of-the-art GDL model. With the consideration of physical validity, this improvement increases to about 300\%.
MolMiner: You only look once for chemical structure recognition
May 23, 2022
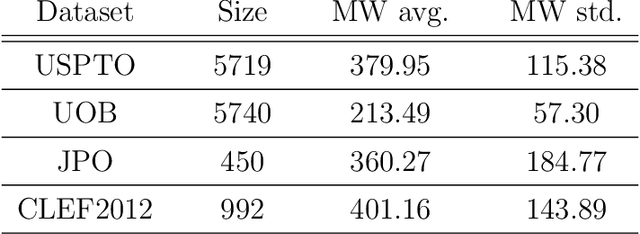
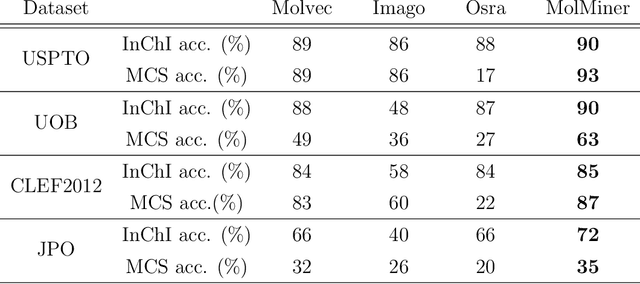
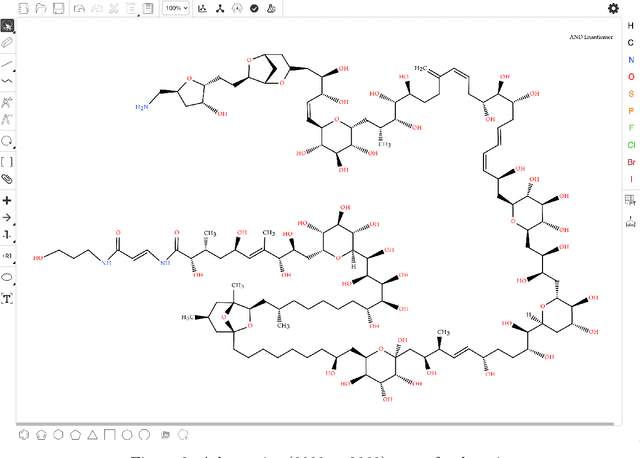
Molecular structures are always depicted as 2D printed form in scientific documents like journal papers and patents. However, these 2D depictions are not machine-readable. Due to a backlog of decades and an increasing amount of these printed literature, there is a high demand for the translation of printed depictions into machine-readable formats, which is known as Optical Chemical Structure Recognition (OCSR). Most OCSR systems developed over the last three decades follow a rule-based approach where the key step of vectorization of the depiction is based on the interpretation of vectors and nodes as bonds and atoms. Here, we present a practical software MolMiner, which is primarily built up using deep neural networks originally developed for semantic segmentation and object detection to recognize atom and bond elements from documents. These recognized elements can be easily connected as a molecular graph with distance-based construction algorithm. We carefully evaluate our software on four benchmark datasets with the state-of-the-art performance. Various real application scenarios are also tested, yielding satisfactory outcomes. The free download links of Mac and Windows versions are available: Mac: https://molminer-cdn.iipharma.cn/pharma-mind/artifact/latest/mac/PharmaMind-mac-latest-setup.dmg and Windows: https://molminer-cdn.iipharma.cn/pharma-mind/artifact/latest/win/PharmaMind-win-latest-setup.exe