 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproved large-scale graph learning through ridge spectral sparsification
Apr 22, 2026Graph-based techniques and spectral graph theory have enriched the field of machine learning with a variety of critical advances. A central object in the analysis is the graph Laplacian L, which encodes the structure of the graph. We consider the problem of learning over this Laplacian in a distributed streaming setting, where new edges of the graph are observed in real time by a network of workers. In this setting, it is hard to learn quickly or approximately while keeping a distributed representation of L. To address this challenge, we present a novel algorithm, GSQUEAK, which efficiently sparsifies the Laplacian by maintaining a small subset of effective resistances. We show that our algorithm produces sparsifiers with strong spectral approximation guarantees, all while processing edges in a single pass and in a distributed fashion.
Hierarchical Mamba Meets Hyperbolic Geometry: A New Paradigm for Structured Language Embeddings
May 25, 2025



Selective state-space models have achieved great success in long-sequence modeling. However, their capacity for language representation, especially in complex hierarchical reasoning tasks, remains underexplored. Most large language models rely on flat Euclidean embeddings, limiting their ability to capture latent hierarchies. To address this limitation, we propose Hierarchical Mamba (HiM), integrating efficient Mamba2 with exponential growth and curved nature of hyperbolic geometry to learn hierarchy-aware language embeddings for deeper linguistic understanding. Mamba2-processed sequences are projected to the Poincare ball (via tangent-based mapping) or Lorentzian manifold (via cosine and sine-based mapping) with "learnable" curvature, optimized with a combined hyperbolic loss. Our HiM model facilitates the capture of relational distances across varying hierarchical levels, enabling effective long-range reasoning. This makes it well-suited for tasks like mixed-hop prediction and multi-hop inference in hierarchical classification. We evaluated our HiM with four linguistic and medical datasets for mixed-hop prediction and multi-hop inference tasks. Experimental results demonstrated that: 1) Both HiM models effectively capture hierarchical relationships for four ontological datasets, surpassing Euclidean baselines. 2) HiM-Poincare captures fine-grained semantic distinctions with higher h-norms, while HiM-Lorentz provides more stable, compact, and hierarchy-preserving embeddings favoring robustness over detail.
Minimum Weighted Feedback Arc Sets for Ranking from Pairwise Comparisons
Dec 10, 2024The Minimum Weighted Feedback Arc Set (MWFAS) problem is fundamentally connected to the Ranking Problem -- the task of deriving global rankings from pairwise comparisons. Recent work [He et al. ICML2022] has advanced the state-of-the-art for the Ranking Problem using learning-based methods, improving upon multiple previous approaches. However, the connection to MWFAS remains underexplored. This paper investigates this relationship and presents efficient combinatorial algorithms for solving MWFAS, thus addressing the Ranking Problem. Our experimental results demonstrate that these simple, learning-free algorithms not only significantly outperform learning-based methods in terms of speed but also generally achieve superior ranking accuracy.
TGB 2.0: A Benchmark for Learning on Temporal Knowledge Graphs and Heterogeneous Graphs
Jun 14, 2024Multi-relational temporal graphs are powerful tools for modeling real-world data, capturing the evolving and interconnected nature of entities over time. Recently, many novel models are proposed for ML on such graphs intensifying the need for robust evaluation and standardized benchmark datasets. However, the availability of such resources remains scarce and evaluation faces added complexity due to reproducibility issues in experimental protocols. To address these challenges, we introduce Temporal Graph Benchmark 2.0 (TGB 2.0), a novel benchmarking framework tailored for evaluating methods for predicting future links on Temporal Knowledge Graphs and Temporal Heterogeneous Graphs with a focus on large-scale datasets, extending the Temporal Graph Benchmark. TGB 2.0 facilitates comprehensive evaluations by presenting eight novel datasets spanning five domains with up to 53 million edges. TGB 2.0 datasets are significantly larger than existing datasets in terms of number of nodes, edges, or timestamps. In addition, TGB 2.0 provides a reproducible and realistic evaluation pipeline for multi-relational temporal graphs. Through extensive experimentation, we observe that 1) leveraging edge-type information is crucial to obtain high performance, 2) simple heuristic baselines are often competitive with more complex methods, 3) most methods fail to run on our largest datasets, highlighting the need for research on more scalable methods.
Resource-constrained knowledge diffusion processes inspired by human peer learning
Dec 01, 2023



We consider a setting where a population of artificial learners is given, and the objective is to optimize aggregate measures of performance, under constraints on training resources. The problem is motivated by the study of peer learning in human educational systems. In this context, we study natural knowledge diffusion processes in networks of interacting artificial learners. By `natural', we mean processes that reflect human peer learning where the students' internal state and learning process is mostly opaque, and the main degree of freedom lies in the formation of peer learning groups by a coordinator who can potentially evaluate the learners before assigning them to peer groups. Among else, we empirically show that such processes indeed make effective use of the training resources, and enable the design of modular neural models that have the capacity to generalize without being prone to overfitting noisy labels.
Towards Foundational Models for Molecular Learning on Large-Scale Multi-Task Datasets
Oct 18, 2023



Recently, pre-trained foundation models have enabled significant advancements in multiple fields. In molecular machine learning, however, where datasets are often hand-curated, and hence typically small, the lack of datasets with labeled features, and codebases to manage those datasets, has hindered the development of foundation models. In this work, we present seven novel datasets categorized by size into three distinct categories: ToyMix, LargeMix and UltraLarge. These datasets push the boundaries in both the scale and the diversity of supervised labels for molecular learning. They cover nearly 100 million molecules and over 3000 sparsely defined tasks, totaling more than 13 billion individual labels of both quantum and biological nature. In comparison, our datasets contain 300 times more data points than the widely used OGB-LSC PCQM4Mv2 dataset, and 13 times more than the quantum-only QM1B dataset. In addition, to support the development of foundational models based on our proposed datasets, we present the Graphium graph machine learning library which simplifies the process of building and training molecular machine learning models for multi-task and multi-level molecular datasets. Finally, we present a range of baseline results as a starting point of multi-task and multi-level training on these datasets. Empirically, we observe that performance on low-resource biological datasets show improvement by also training on large amounts of quantum data. This indicates that there may be potential in multi-task and multi-level training of a foundation model and fine-tuning it to resource-constrained downstream tasks.
Neural Network Pruning as Spectrum Preserving Process
Jul 18, 2023



Neural networks have achieved remarkable performance in various application domains. Nevertheless, a large number of weights in pre-trained deep neural networks prohibit them from being deployed on smartphones and embedded systems. It is highly desirable to obtain lightweight versions of neural networks for inference in edge devices. Many cost-effective approaches were proposed to prune dense and convolutional layers that are common in deep neural networks and dominant in the parameter space. However, a unified theoretical foundation for the problem mostly is missing. In this paper, we identify the close connection between matrix spectrum learning and neural network training for dense and convolutional layers and argue that weight pruning is essentially a matrix sparsification process to preserve the spectrum. Based on the analysis, we also propose a matrix sparsification algorithm tailored for neural network pruning that yields better pruning result. We carefully design and conduct experiments to support our arguments. Hence we provide a consolidated viewpoint for neural network pruning and enhance the interpretability of deep neural networks by identifying and preserving the critical neural weights.
K-SpecPart: A Supervised Spectral Framework for Multi-Way Hypergraph Partitioning Solution Improvement
May 07, 2023



State-of-the-art hypergraph partitioners follow the multilevel paradigm, constructing multiple levels of coarser hypergraphs to drive cutsize refinement. These partitioners face limitations: (i) coarsening processes depend on local neighborhood structure, ignoring global hypergraph structure; (ii) refinement heuristics risk entrapment in local minima. We introduce K-SpecPart, a supervised spectral framework addressing these limitations by solving a generalized eigenvalue problem, capturing balanced partitioning objectives and global hypergraph structure in a low-dimensional vertex embedding while leveraging high-quality multilevel partitioning solutions as hints. In multi-way partitioning, K-SpecPart derives multiple bipartitioning solutions from a multi-way hint partitioning solution. It integrates these solutions into the generalized eigenvalue problem to compute eigenvectors, creating a large-dimensional embedding. Linear Discriminant Analysis (LDA) is used to transform this into a lower-dimensional embedding. K-SpecPart constructs a family of trees from the vertex embedding and partitions them using a tree-sweeping algorithm. We extend SpecPart's tree partitioning algorithm for multi-way partitioning. The multiple tree-based partitioning solutions are overlaid, followed by lifting to a clustered hypergraph where an integer linear programming (ILP) partitioning problem is solved. Empirical studies show K-SpecPart's benefits. For bipartitioning, K-SpecPart outperforms SpecPart with improvements up to 30%. For multi-way partitioning, K-SpecPart surpasses hMETIS and KaHyPar, with improvements up to 20% in some cases.
SGC: A semi-supervised pipeline for gene clustering using self-training approach in gene co-expression networks
Sep 21, 2022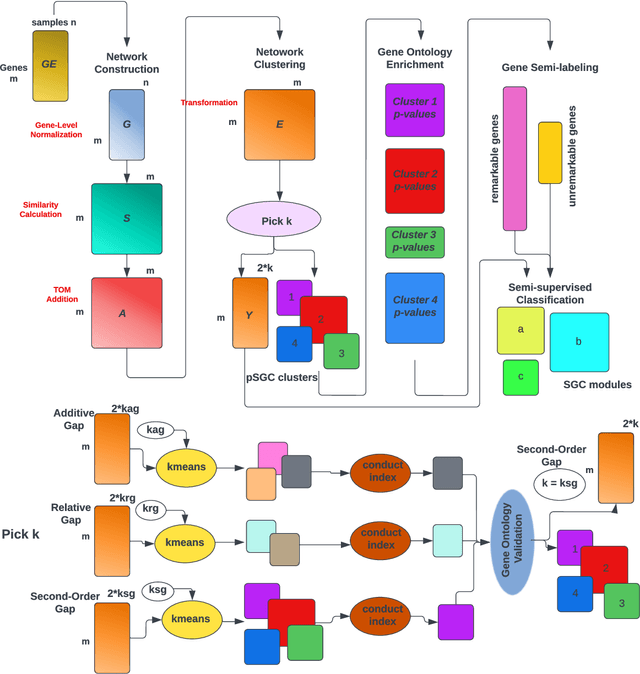
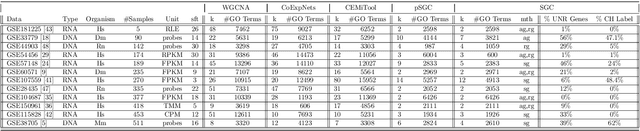
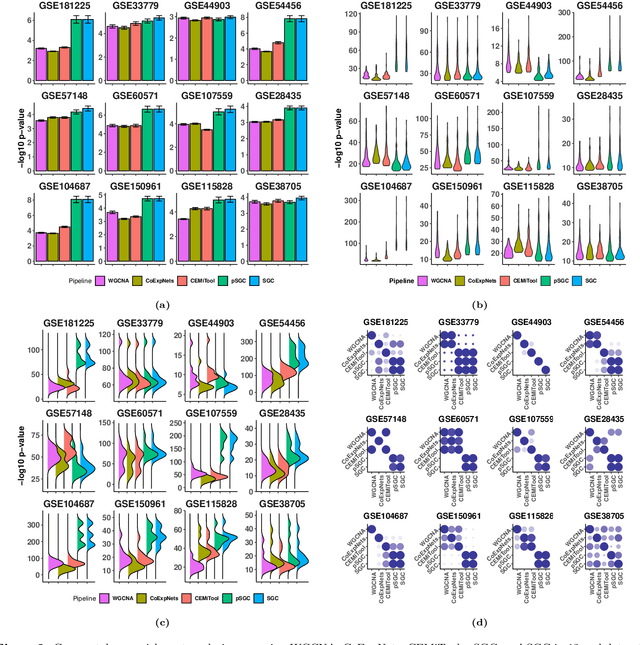
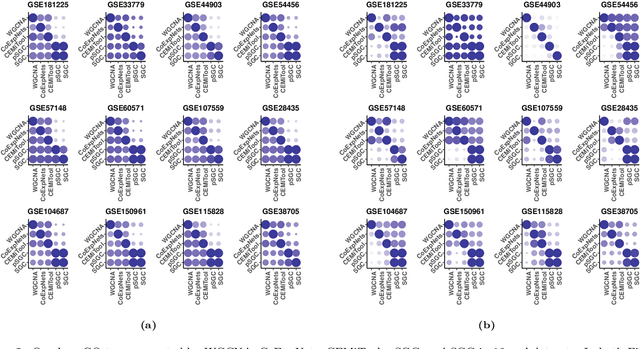
A widely used approach for extracting information from gene expression data employ the construction of a gene co-expression network and the subsequent application of algorithms that discover network structure. In particular, a common goal is the computational discovery of gene clusters, commonly called modules. When applied on a novel gene expression dataset, the quality of the computed modules can be evaluated automatically, using Gene Ontology enrichment, a method that measures the frequencies of Gene Ontology terms in the computed modules and evaluates their statistical likelihood. In this work we propose SGC a novel pipeline for gene clustering based on relatively recent seminal work in the mathematics of spectral network theory. SGC consists of multiple novel steps that enable the computation of highly enriched modules in an unsupervised manner. But unlike all existing frameworks, it further incorporates a novel step that leverages Gene Ontology information in a semi-supervised clustering method that further improves the quality of the computed modules. Comparing with already well-known existing frameworks, we show that SGC results in higher enrichment in real data. In particular, in 12 real gene expression datasets, SGC outperforms in all except one.
Incremental Spectral Sparsification for Large-Scale Graph-Based Semi-Supervised Learning
Jan 21, 2016


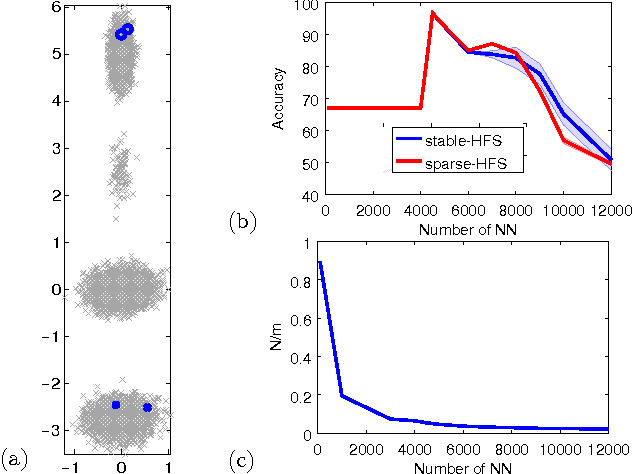
While the harmonic function solution performs well in many semi-supervised learning (SSL) tasks, it is known to scale poorly with the number of samples. Recent successful and scalable methods, such as the eigenfunction method focus on efficiently approximating the whole spectrum of the graph Laplacian constructed from the data. This is in contrast to various subsampling and quantization methods proposed in the past, which may fail in preserving the graph spectra. However, the impact of the approximation of the spectrum on the final generalization error is either unknown, or requires strong assumptions on the data. In this paper, we introduce Sparse-HFS, an efficient edge-sparsification algorithm for SSL. By constructing an edge-sparse and spectrally similar graph, we are able to leverage the approximation guarantees of spectral sparsification methods to bound the generalization error of Sparse-HFS. As a result, we obtain a theoretically-grounded approximation scheme for graph-based SSL that also empirically matches the performance of known large-scale methods.