 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMultivariate Stochastic Dominance via Optimal Transport and Applications to Models Benchmarking
Jun 10, 2024Stochastic dominance is an important concept in probability theory, econometrics and social choice theory for robustly modeling agents' preferences between random outcomes. While many works have been dedicated to the univariate case, little has been done in the multivariate scenario, wherein an agent has to decide between different multivariate outcomes. By exploiting a characterization of multivariate first stochastic dominance in terms of couplings, we introduce a statistic that assesses multivariate almost stochastic dominance under the framework of Optimal Transport with a smooth cost. Further, we introduce an entropic regularization of this statistic, and establish a central limit theorem (CLT) and consistency of the bootstrap procedure for the empirical statistic. Armed with this CLT, we propose a hypothesis testing framework as well as an efficient implementation using the Sinkhorn algorithm. We showcase our method in comparing and benchmarking Large Language Models that are evaluated on multiple metrics. Our multivariate stochastic dominance test allows us to capture the dependencies between the metrics in order to make an informed and statistically significant decision on the relative performance of the models.
Information Theoretic Guarantees For Policy Alignment In Large Language Models
Jun 09, 2024

Policy alignment of large language models refers to constrained policy optimization, where the policy is optimized to maximize a reward while staying close to a reference policy with respect to an $f$-divergence such as the $\mathsf{KL}$ divergence. The best of $n$ alignment policy selects a sample from the reference policy that has the maximum reward among $n$ independent samples. For both cases (policy alignment and best of $n$), recent works showed empirically that the reward improvement of the aligned policy on the reference one scales like $\sqrt{\mathsf{KL}}$, with an explicit bound in $n$ on the $\mathsf{KL}$ for the best of $n$ policy. We show in this paper that the $\sqrt{\mathsf{KL}}$ information theoretic upper bound holds if the reward under the reference policy has sub-gaussian tails. Moreover, we prove for the best of $n$ policy, that the $\mathsf{KL}$ upper bound can be obtained for any $f$-divergence via a reduction to exponential order statistics owing to the R\'enyi representation of order statistics, and a data processing inequality. If additional information is known on the tails of the aligned policy we show that tighter control on the reward improvement can be obtained via the R\'enyi divergence. Finally we demonstrate how these upper bounds transfer from proxy rewards to golden rewards which results in a decrease in the golden reward improvement due to overestimation and approximation errors of the proxy reward.
Distributional Preference Alignment of LLMs via Optimal Transport
Jun 09, 2024



Current LLM alignment techniques use pairwise human preferences at a sample level, and as such, they do not imply an alignment on the distributional level. We propose in this paper Alignment via Optimal Transport (AOT), a novel method for distributional preference alignment of LLMs. AOT aligns LLMs on unpaired preference data by making the reward distribution of the positive samples stochastically dominant in the first order on the distribution of negative samples. We introduce a convex relaxation of this first-order stochastic dominance and cast it as an optimal transport problem with a smooth and convex cost. Thanks to the one-dimensional nature of the resulting optimal transport problem and the convexity of the cost, it has a closed-form solution via sorting on empirical measures. We fine-tune LLMs with this AOT objective, which enables alignment by penalizing the violation of the stochastic dominance of the reward distribution of the positive samples on the reward distribution of the negative samples. We analyze the sample complexity of AOT by considering the dual of the OT problem and show that it converges at the parametric rate. Empirically, we show on a diverse set of alignment datasets and LLMs that AOT leads to state-of-the-art models in the 7B family of models when evaluated with Open LLM Benchmarks and AlpacaEval.
Risk Assessment and Statistical Significance in the Age of Foundation Models
Oct 11, 2023



We propose a distributional framework for assessing socio-technical risks of foundation models with quantified statistical significance. Our approach hinges on a new statistical relative testing based on first and second order stochastic dominance of real random variables. We show that the second order statistics in this test are linked to mean-risk models commonly used in econometrics and mathematical finance to balance risk and utility when choosing between alternatives. Using this framework, we formally develop a risk-aware approach for foundation model selection given guardrails quantified by specified metrics. Inspired by portfolio optimization and selection theory in mathematical finance, we define a \emph{metrics portfolio} for each model as a means to aggregate a collection of metrics, and perform model selection based on the stochastic dominance of these portfolios. The statistical significance of our tests is backed theoretically by an asymptotic analysis via central limit theorems instantiated in practice via a bootstrap variance estimate. We use our framework to compare various large language models regarding risks related to drifting from instructions and outputting toxic content.
Auditing and Generating Synthetic Data with Controllable Trust Trade-offs
May 02, 2023



Data collected from the real world tends to be biased, unbalanced, and at risk of exposing sensitive and private information. This reality has given rise to the idea of creating synthetic datasets to alleviate risk, bias, harm, and privacy concerns inherent in the real data. This concept relies on Generative AI models to produce unbiased, privacy-preserving synthetic data while being true to the real data. In this new paradigm, how can we tell if this approach delivers on its promises? We present an auditing framework that offers a holistic assessment of synthetic datasets and AI models trained on them, centered around bias and discrimination prevention, fidelity to the real data, utility, robustness, and privacy preservation. We showcase our framework by auditing multiple generative models on diverse use cases, including education, healthcare, banking, human resources, and across different modalities, from tabular, to time-series, to natural language. Our use cases demonstrate the importance of a holistic assessment in order to ensure compliance with socio-technical safeguards that regulators and policymakers are increasingly enforcing. For this purpose, we introduce the trust index that ranks multiple synthetic datasets based on their prescribed safeguards and their desired trade-offs. Moreover, we devise a trust-index-driven model selection and cross-validation procedure via auditing in the training loop that we showcase on a class of transformer models that we dub TrustFormers, across different modalities. This trust-driven model selection allows for controllable trust trade-offs in the resulting synthetic data. We instrument our auditing framework with workflows that connect different stakeholders from model development to audit and certification via a synthetic data auditing report.
Effective Dynamics of Generative Adversarial Networks
Dec 08, 2022Generative adversarial networks (GANs) are a class of machine-learning models that use adversarial training to generate new samples with the same (potentially very complex) statistics as the training samples. One major form of training failure, known as mode collapse, involves the generator failing to reproduce the full diversity of modes in the target probability distribution. Here, we present an effective model of GAN training, which captures the learning dynamics by replacing the generator neural network with a collection of particles in the output space; particles are coupled by a universal kernel valid for certain wide neural networks and high-dimensional inputs. The generality of our simplified model allows us to study the conditions under which mode collapse occurs. Indeed, experiments which vary the effective kernel of the generator reveal a mode collapse transition, the shape of which can be related to the type of discriminator through the frequency principle. Further, we find that gradient regularizers of intermediate strengths can optimally yield convergence through critical damping of the generator dynamics. Our effective GAN model thus provides an interpretable physical framework for understanding and improving adversarial training.
Cloud-Based Real-Time Molecular Screening Platform with MolFormer
Aug 13, 2022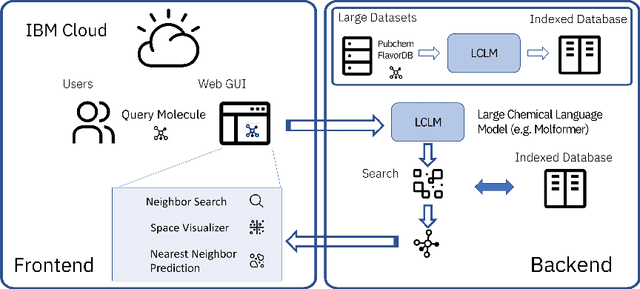
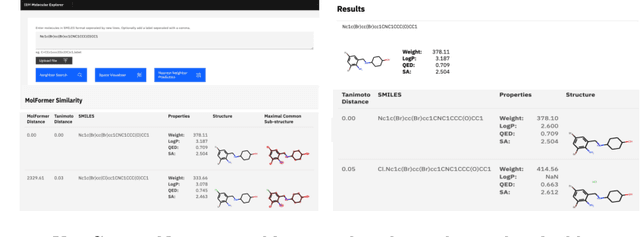
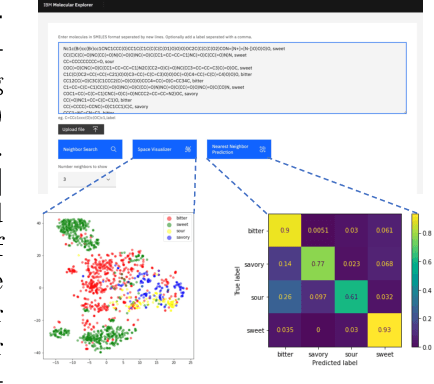
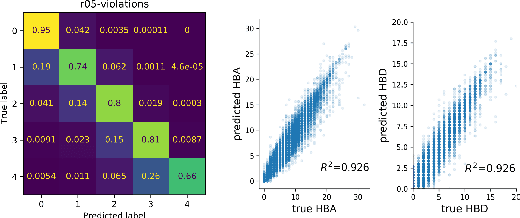
With the prospect of automating a number of chemical tasks with high fidelity, chemical language processing models are emerging at a rapid speed. Here, we present a cloud-based real-time platform that allows users to virtually screen molecules of interest. For this purpose, molecular embeddings inferred from a recently proposed large chemical language model, named MolFormer, are leveraged. The platform currently supports three tasks: nearest neighbor retrieval, chemical space visualization, and property prediction. Based on the functionalities of this platform and results obtained, we believe that such a platform can play a pivotal role in automating chemistry and chemical engineering research, as well as assist in drug discovery and material design tasks. A demo of our platform is provided at \url{www.ibm.biz/molecular_demo}.
Auditing Differential Privacy in High Dimensions with the Kernel Quantum Rényi Divergence
May 27, 2022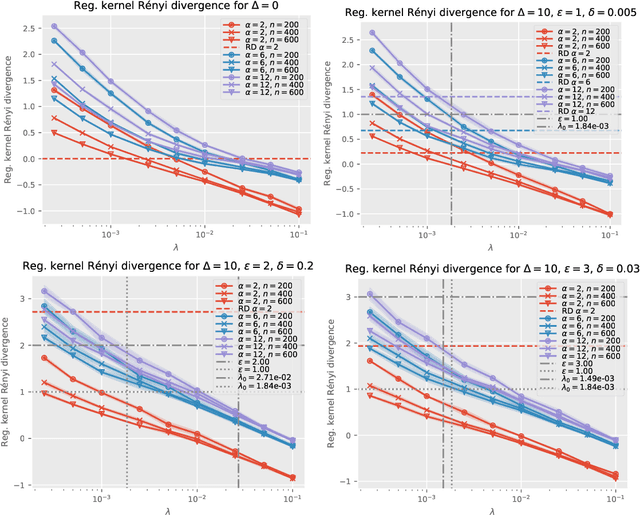
Differential privacy (DP) is the de facto standard for private data release and private machine learning. Auditing black-box DP algorithms and mechanisms to certify whether they satisfy a certain DP guarantee is challenging, especially in high dimension. We propose relaxations of differential privacy based on new divergences on probability distributions: the kernel R\'enyi divergence and its regularized version. We show that the regularized kernel R\'enyi divergence can be estimated from samples even in high dimensions, giving rise to auditing procedures for $\varepsilon$-DP, $(\varepsilon,\delta)$-DP and $(\alpha,\varepsilon)$-R\'enyi DP.
Learning with Stochastic Orders
May 27, 2022
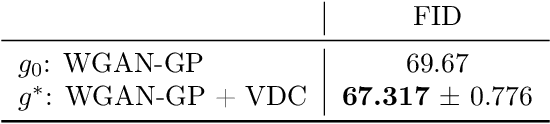

Learning high-dimensional distributions is often done with explicit likelihood modeling or implicit modeling via minimizing integral probability metrics (IPMs). In this paper, we expand this learning paradigm to stochastic orders, namely, the convex or Choquet order between probability measures. Towards this end, we introduce the Choquet-Toland distance between probability measures, that can be used as a drop-in replacement for IPMs. We also introduce the Variational Dominance Criterion (VDC) to learn probability measures with dominance constraints, that encode the desired stochastic order between the learned measure and a known baseline. We analyze both quantities and show that they suffer from the curse of dimensionality and propose surrogates via input convex maxout networks (ICMNs), that enjoy parametric rates. Finally, we provide a min-max framework for learning with stochastic orders and validate it experimentally on synthetic and high-dimensional image generation, with promising results. The code is available at https://github.com/yair-schiff/stochastic-orders-ICMN
Cycle Consistent Probability Divergences Across Different Spaces
Nov 22, 2021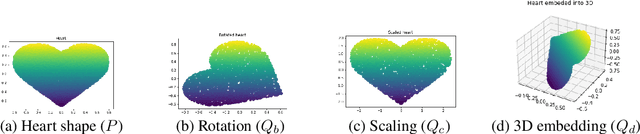
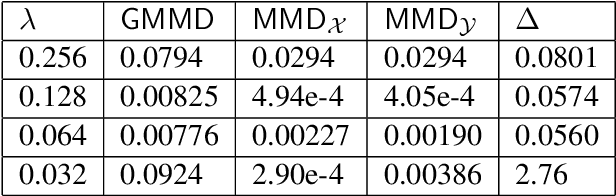
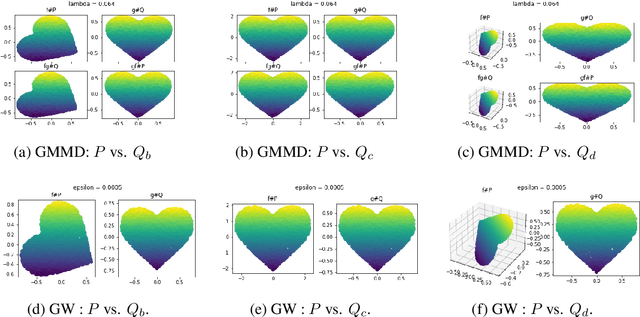
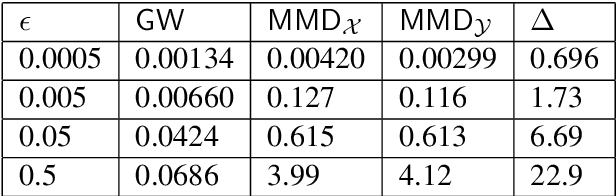
Discrepancy measures between probability distributions are at the core of statistical inference and machine learning. In many applications, distributions of interest are supported on different spaces, and yet a meaningful correspondence between data points is desired. Motivated to explicitly encode consistent bidirectional maps into the discrepancy measure, this work proposes a novel unbalanced Monge optimal transport formulation for matching, up to isometries, distributions on different spaces. Our formulation arises as a principled relaxation of the Gromov-Haussdroff distance between metric spaces, and employs two cycle-consistent maps that push forward each distribution onto the other. We study structural properties of the proposed discrepancy and, in particular, show that it captures the popular cycle-consistent generative adversarial network (GAN) framework as a special case, thereby providing the theory to explain it. Motivated by computational efficiency, we then kernelize the discrepancy and restrict the mappings to parametric function classes. The resulting kernelized version is coined the generalized maximum mean discrepancy (GMMD). Convergence rates for empirical estimation of GMMD are studied and experiments to support our theory are provided.