 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTargeting SARS-CoV-2 with AI- and HPC-enabled Lead Generation: A First Data Release
May 28, 2020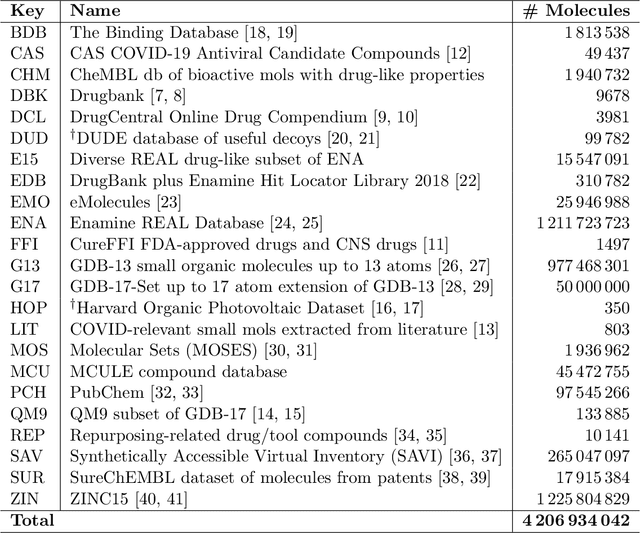
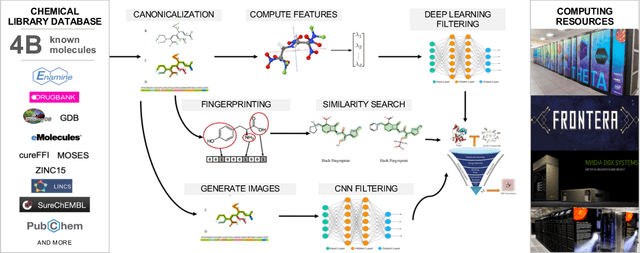

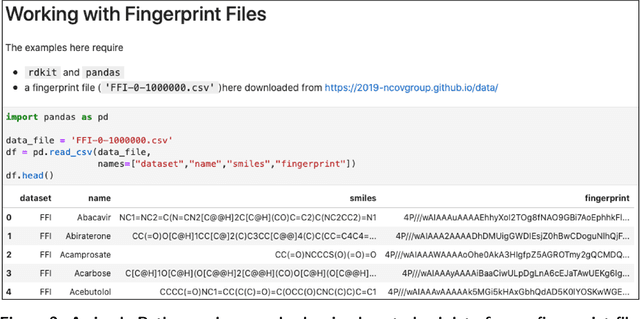
Researchers across the globe are seeking to rapidly repurpose existing drugs or discover new drugs to counter the the novel coronavirus disease (COVID-19) caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). One promising approach is to train machine learning (ML) and artificial intelligence (AI) tools to screen large numbers of small molecules. As a contribution to that effort, we are aggregating numerous small molecules from a variety of sources, using high-performance computing (HPC) to computer diverse properties of those molecules, using the computed properties to train ML/AI models, and then using the resulting models for screening. In this first data release, we make available 23 datasets collected from community sources representing over 4.2 B molecules enriched with pre-computed: 1) molecular fingerprints to aid similarity searches, 2) 2D images of molecules to enable exploration and application of image-based deep learning methods, and 3) 2D and 3D molecular descriptors to speed development of machine learning models. This data release encompasses structural information on the 4.2 B molecules and 60 TB of pre-computed data. Future releases will expand the data to include more detailed molecular simulations, computed models, and other products.
Learning Everywhere: A Taxonomy for the Integration of Machine Learning and Simulations
Oct 13, 2019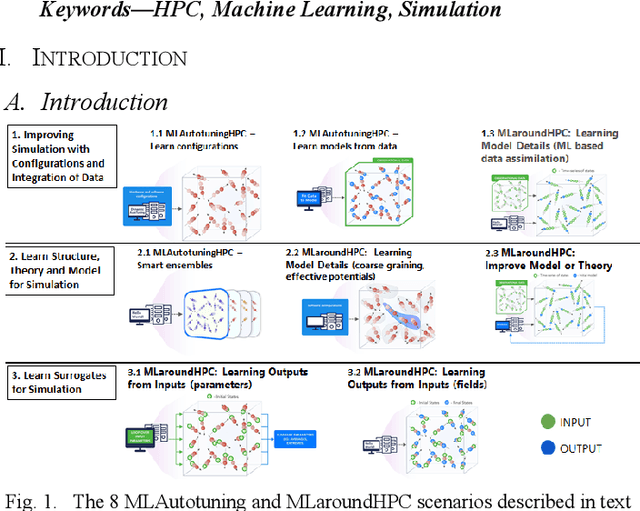
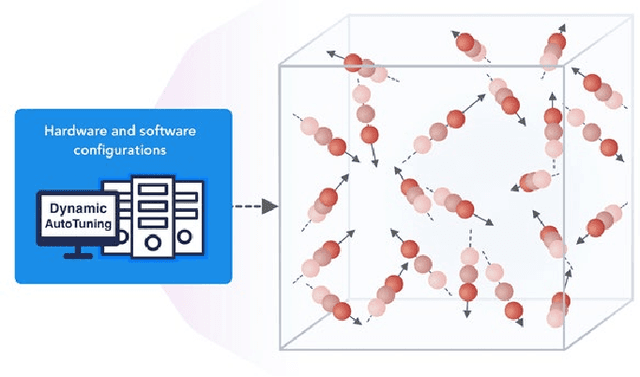
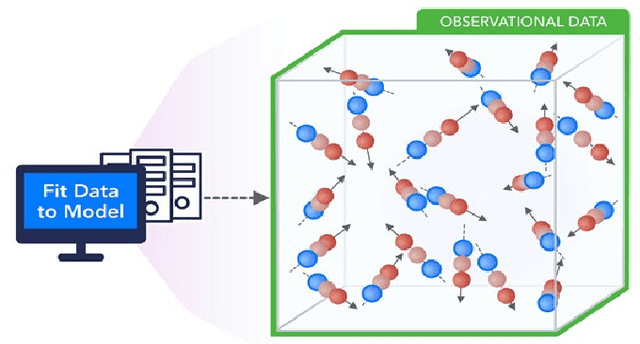
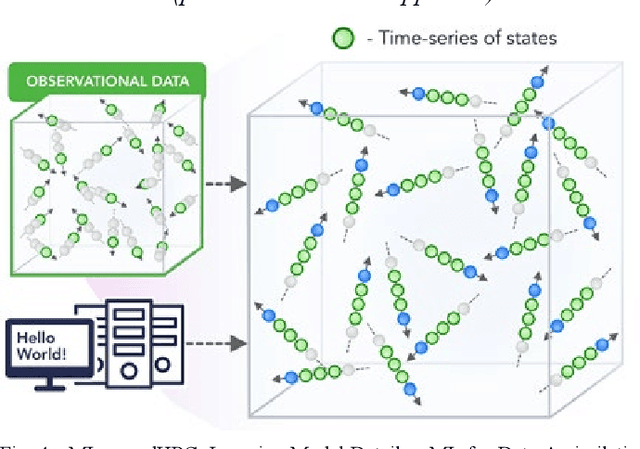
We present a taxonomy of research on Machine Learning (ML) applied to enhance simulations together with a catalog of some activities. We cover eight patterns for the link of ML to the simulations or systems plus three algorithmic areas: particle dynamics, agent-based models and partial differential equations. The patterns are further divided into three action areas: Improving simulation with Configurations and Integration of Data, Learn Structure, Theory and Model for Simulation, and Learn to make Surrogates.
DeepDriveMD: Deep-Learning Driven Adaptive Molecular Simulations for Protein Folding
Sep 17, 2019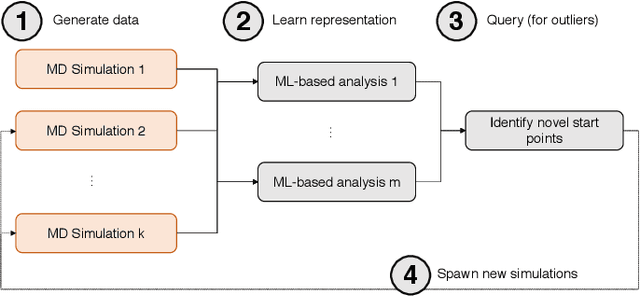
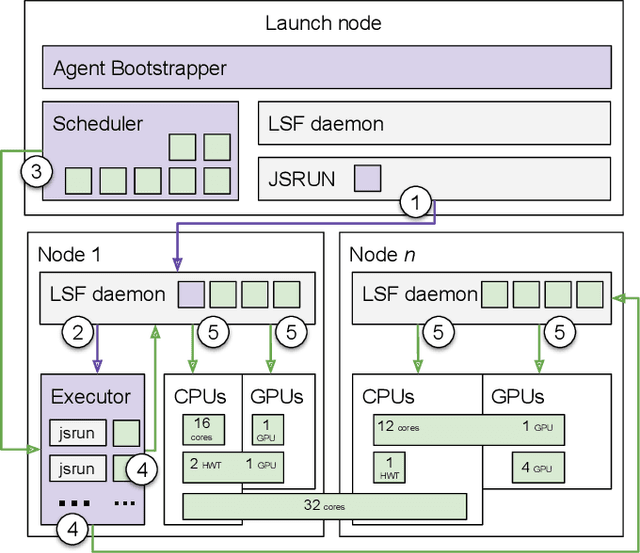
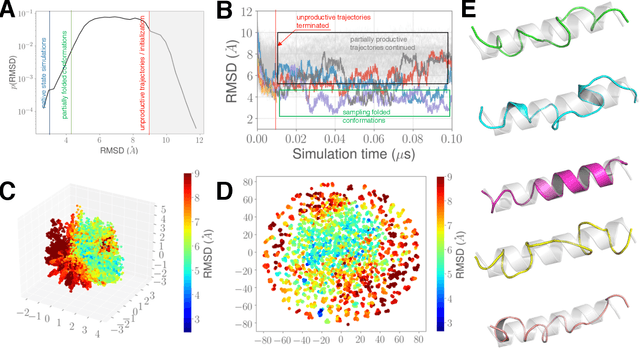
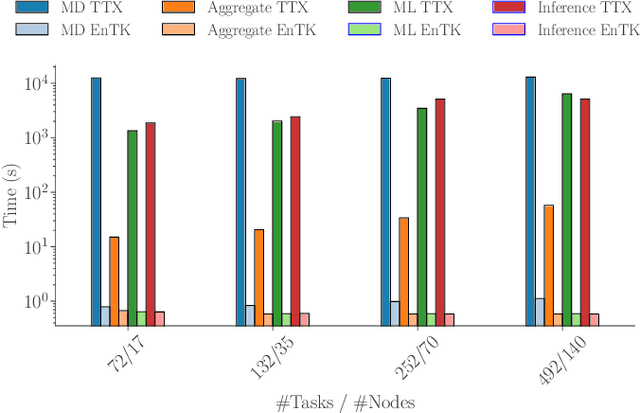
Simulations of biological macromolecules play an important role in understanding the physical basis of a number of complex processes such as protein folding. Even with increasing computational power and evolution of specialized architectures, the ability to simulate protein folding at atomistic scales still remains challenging. This stems from the dual aspects of high dimensionality of protein conformational landscapes, and the inability of atomistic molecular dynamics (MD) simulations to sufficiently sample these landscapes to observe folding events. Machine learning/deep learning (ML/DL) techniques, when combined with atomistic MD simulations offer the opportunity to potentially overcome these limitations by: (1) effectively reducing the dimensionality of MD simulations to automatically build latent representations that correspond to biophysically relevant reaction coordinates (RCs), and (2) driving MD simulations to automatically sample potentially novel conformational states based on these RCs. We examine how coupling DL approaches with MD simulations can fold small proteins effectively on supercomputers. In particular, we study the computational costs and effectiveness of scaling DL-coupled MD workflows by folding two prototypical systems, viz., Fs-peptide and the fast-folding variant of the villin head piece protein. We demonstrate that a DL driven MD workflow is able to effectively learn latent representations and drive adaptive simulations. Compared to traditional MD-based approaches, our approach achieves an effective performance gain in sampling the folded states by at least 2.3x. Our study provides a quantitative basis to understand how DL driven MD simulations, can lead to effective performance gains and reduced times to solution on supercomputing resources.
Understanding ML driven HPC: Applications and Infrastructure
Sep 05, 2019We recently outlined the vision of "Learning Everywhere" which captures the possibility and impact of how learning methods and traditional HPC methods can be coupled together. A primary driver of such coupling is the promise that Machine Learning (ML) will give major performance improvements for traditional HPC simulations. Motivated by this potential, the ML around HPC class of integration is of particular significance. In a related follow-up paper, we provided an initial taxonomy for integrating learning around HPC methods. In this paper, which is part of the Learning Everywhere series, we discuss "how" learning methods and HPC simulations are being integrated to enhance effective performance of computations. This paper identifies several modes --- substitution, assimilation, and control, in which learning methods integrate with HPC simulations and provide representative applications in each mode. This paper discusses some open research questions and we hope will motivate and clear the ground for MLaroundHPC benchmarks.
Learning Neural Markers of Schizophrenia Disorder Using Recurrent Neural Networks
Dec 01, 2017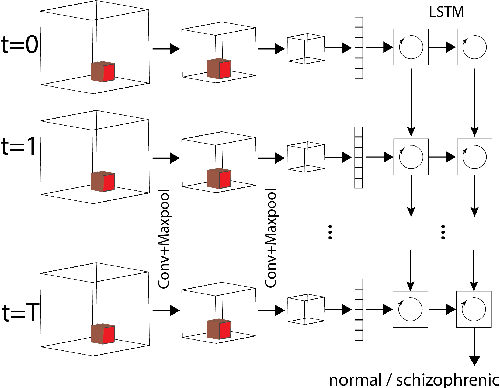
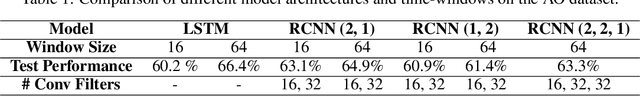
Smart systems that can accurately diagnose patients with mental disorders and identify effective treatments based on brain functional imaging data are of great applicability and are gaining much attention. Most previous machine learning studies use hand-designed features, such as functional connectivity, which does not maintain the potential useful information in the spatial relationship between brain regions and the temporal profile of the signal in each region. Here we propose a new method based on recurrent-convolutional neural networks to automatically learn useful representations from segments of 4-D fMRI recordings. Our goal is to exploit both spatial and temporal information in the functional MRI movie (at the whole-brain voxel level) for identifying patients with schizophrenia.