 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeBehaviour-conditioned policies for cooperative reinforcement learning tasks
Oct 04, 2021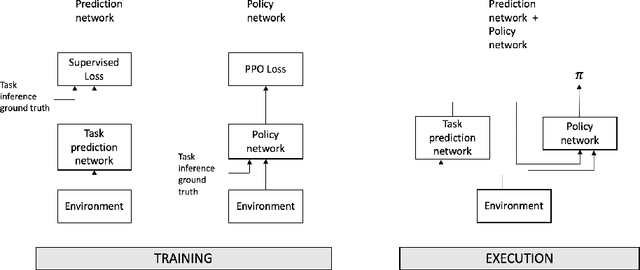

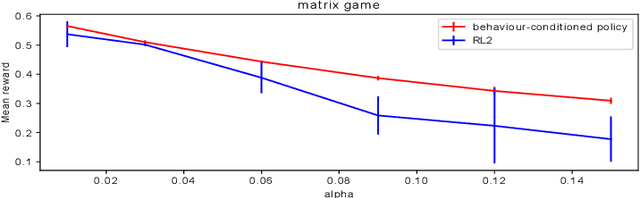
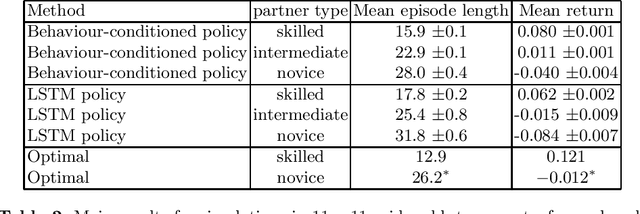
The cooperation among AI systems, and between AI systems and humans is becoming increasingly important. In various real-world tasks, an agent needs to cooperate with unknown partner agent types. This requires the agent to assess the behaviour of the partner agent during a cooperative task and to adjust its own policy to support the cooperation. Deep reinforcement learning models can be trained to deliver the required functionality but are known to suffer from sample inefficiency and slow learning. However, adapting to a partner agent behaviour during the ongoing task requires ability to assess the partner agent type quickly. We suggest a method, where we synthetically produce populations of agents with different behavioural patterns together with ground truth data of their behaviour, and use this data for training a meta-learner. We additionally suggest an agent architecture, which can efficiently use the generated data and gain the meta-learning capability. When an agent is equipped with such a meta-learner, it is capable of quickly adapting to cooperation with unknown partner agent types in new situations. This method can be used to automatically form a task distribution for meta-training from emerging behaviours that arise, for example, through self-play.
Stochastic Cluster Embedding
Aug 18, 2021

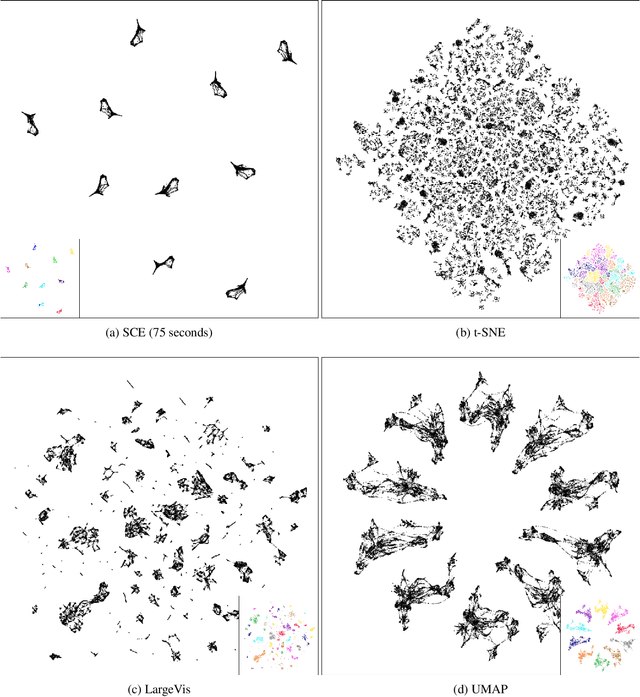
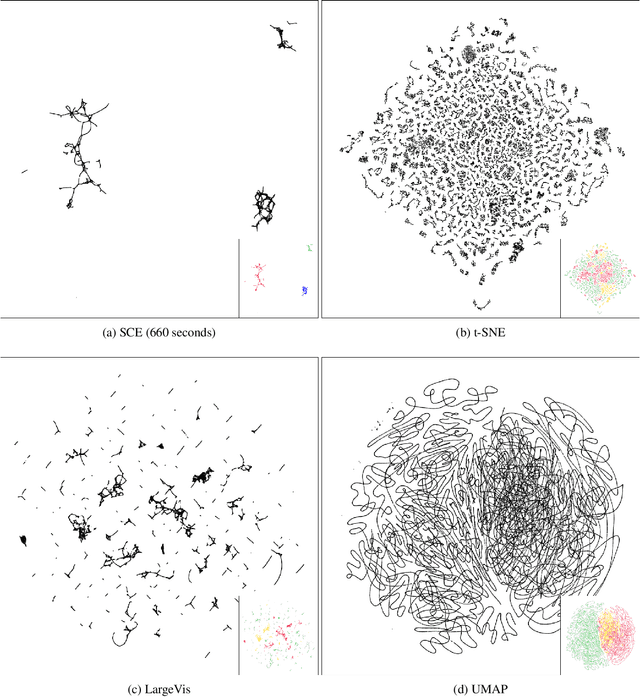
Neighbor Embedding (NE) that aims to preserve pairwise similarities between data items has been shown to yield an effective principle for data visualization. However, even the currently best NE methods such as Stochastic Neighbor Embedding (SNE) may leave large-scale patterns such as clusters hidden despite of strong signals being present in the data. To address this, we propose a new cluster visualization method based on Neighbor Embedding. We first present a family of Neighbor Embedding methods which generalizes SNE by using non-normalized Kullback-Leibler divergence with a scale parameter. In this family, much better cluster visualizations often appear with a parameter value different from the one corresponding to SNE. We also develop an efficient software which employs asynchronous stochastic block coordinate descent to optimize the new family of objective functions. The experimental results demonstrate that our method consistently and substantially improves visualization of data clusters compared with the state-of-the-art NE approaches.
Toward AI Assistants That Let Designers Design
Jul 22, 2021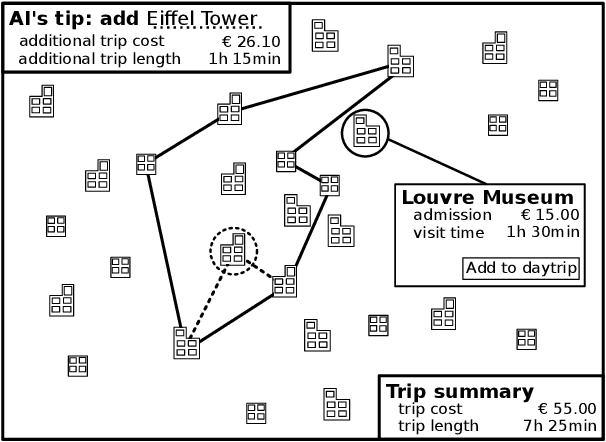
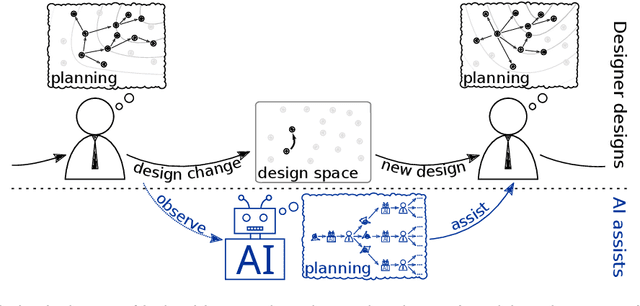
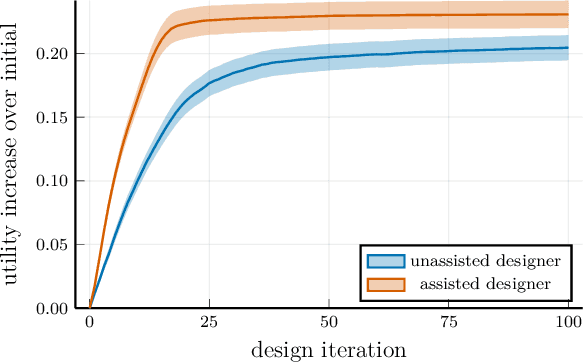
AI for supporting designers needs to be rethought. It should aim to cooperate, not automate, by supporting and leveraging the creativity and problem-solving of designers. The challenge for such AI is how to infer designers' goals and then help them without being needlessly disruptive. We present AI-assisted design: a framework for creating such AI, built around generative user models which enable reasoning about designers' goals, reasoning, and capabilities.
Bayesian inference of ODEs with Gaussian processes
Jun 21, 2021
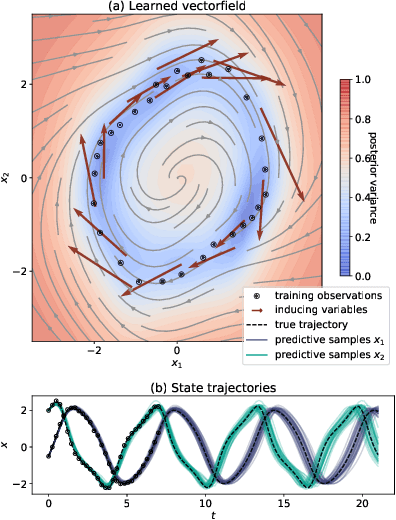
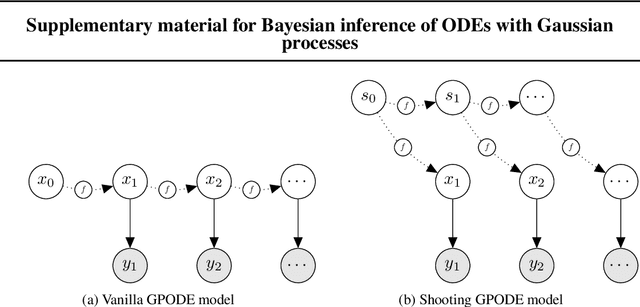
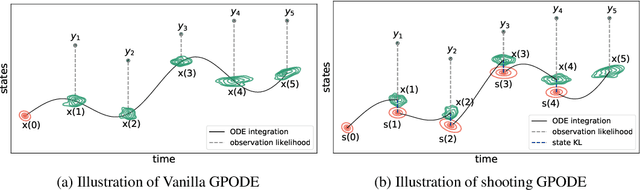
Recent machine learning advances have proposed black-box estimation of unknown continuous-time system dynamics directly from data. However, earlier works are based on approximative ODE solutions or point estimates. We propose a novel Bayesian nonparametric model that uses Gaussian processes to infer posteriors of unknown ODE systems directly from data. We derive sparse variational inference with decoupled functional sampling to represent vector field posteriors. We also introduce a probabilistic shooting augmentation to enable efficient inference from arbitrarily long trajectories. The method demonstrates the benefit of computing vector field posteriors, with predictive uncertainty scores outperforming alternative methods on multiple ODE learning tasks.
Targeted Active Learning for Bayesian Decision-Making
Jun 08, 2021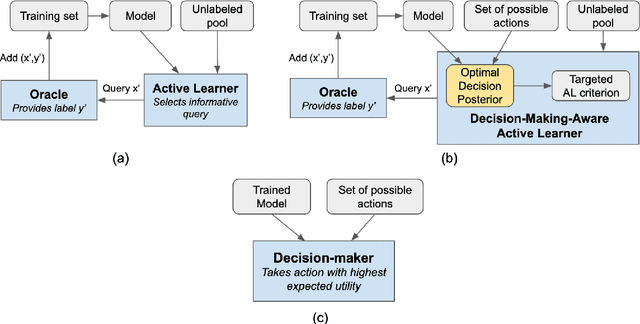
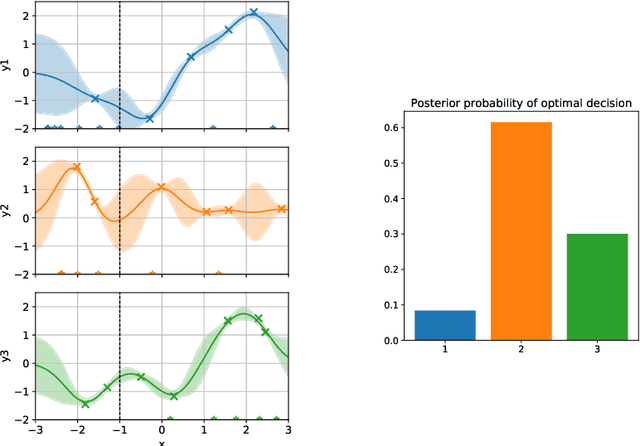
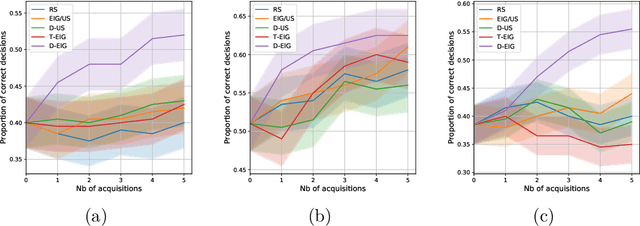
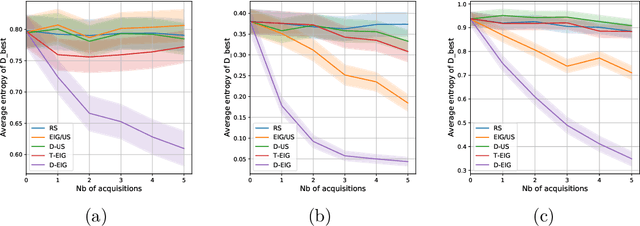
Active learning is usually applied to acquire labels of informative data points in supervised learning, to maximize accuracy in a sample-efficient way. However, maximizing the accuracy is not the end goal when the results are used for decision-making, for example in personalized medicine or economics. We argue that when acquiring samples sequentially, separating learning and decision-making is sub-optimal, and we introduce a novel active learning strategy which takes the down-the-line decision problem into account. Specifically, we introduce a novel active learning criterion which maximizes the expected information gain on the posterior distribution of the optimal decision. We compare our decision-making-aware active learning strategy to existing alternatives on both simulated and real data, and show improved performance in decision-making accuracy.
Affine Transport for Sim-to-Real Domain Adaptation
May 25, 2021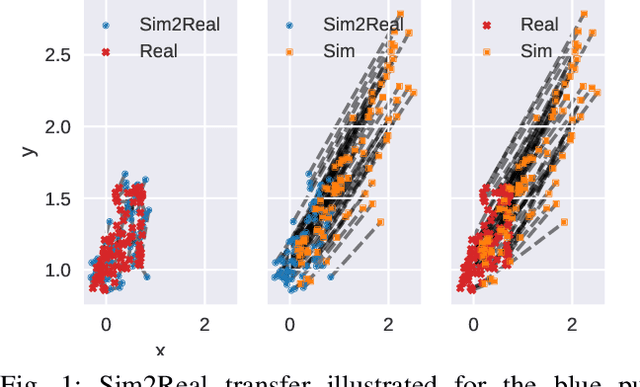

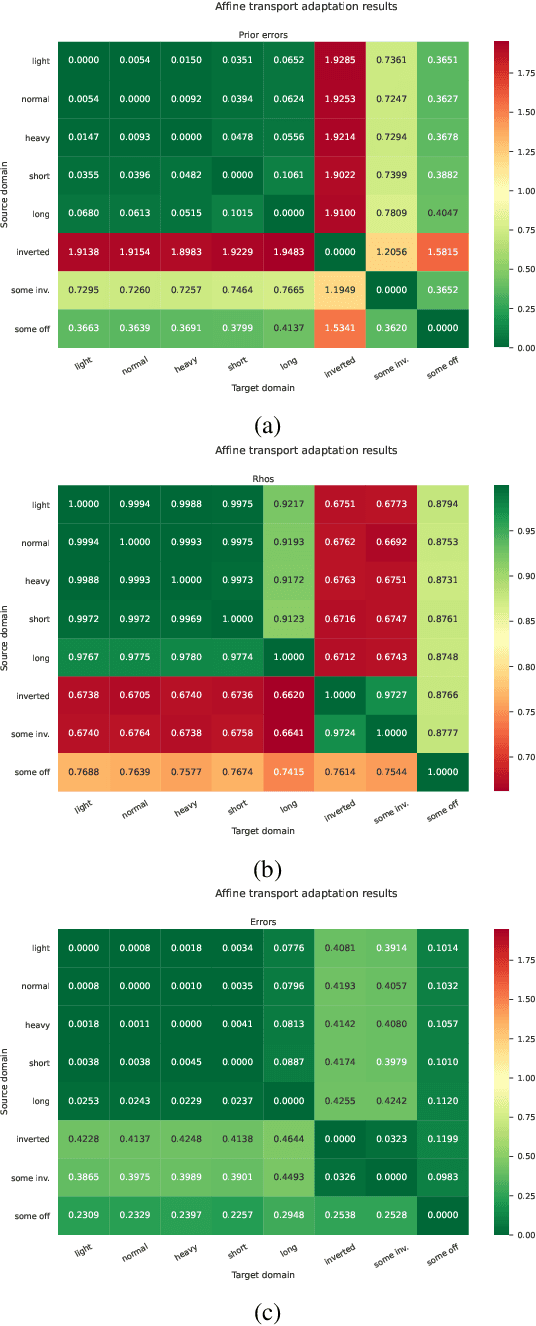
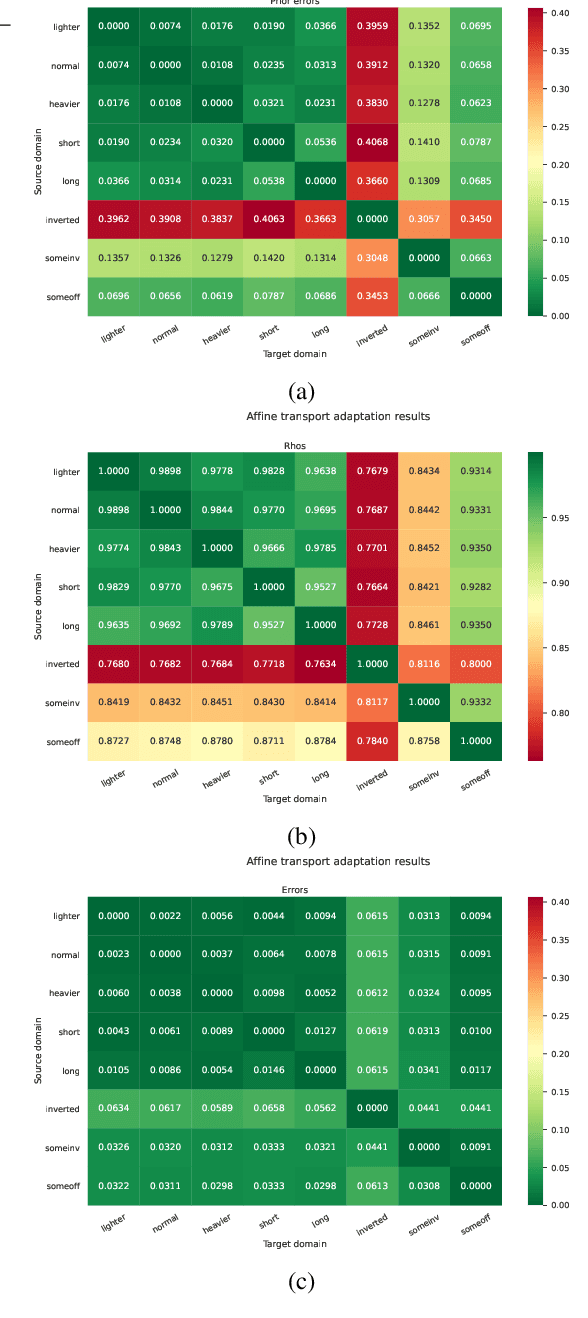
Sample-efficient domain adaptation is an open problem in robotics. In this paper, we present affine transport -- a variant of optimal transport, which models the mapping between state transition distributions between the source and target domains with an affine transformation. First, we derive the affine transport framework; then, we extend the basic framework with Procrustes alignment to model arbitrary affine transformations. We evaluate the method in a number of OpenAI Gym sim-to-sim experiments with simulation environments, as well as on a sim-to-real domain adaptation task of a robot hitting a hockeypuck such that it slides and stops at a target position. In each experiment, we evaluate the results when transferring between each pair of dynamics domains. The results show that affine transport can significantly reduce the model adaptation error in comparison to using the original, non-adapted dynamics model.
DIVERSE: bayesian Data IntegratiVE learning for precise drug ResponSE prediction
Mar 31, 2021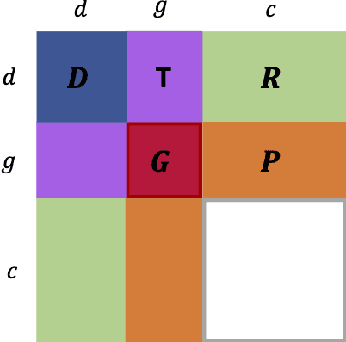
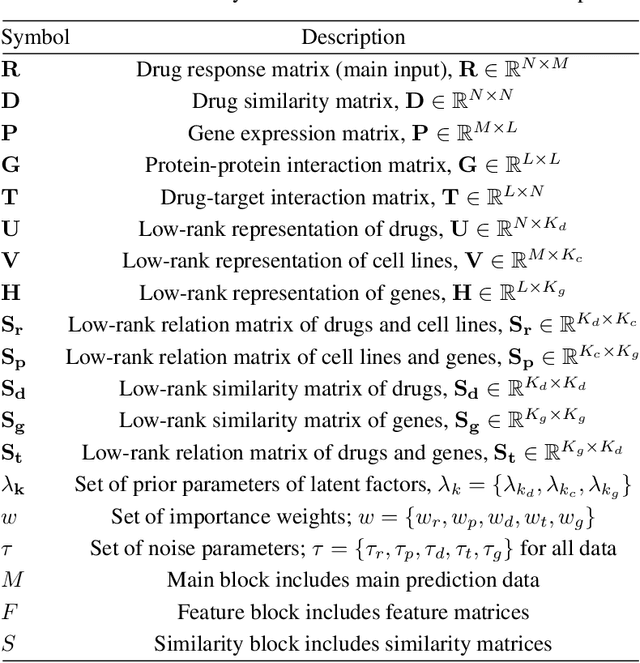
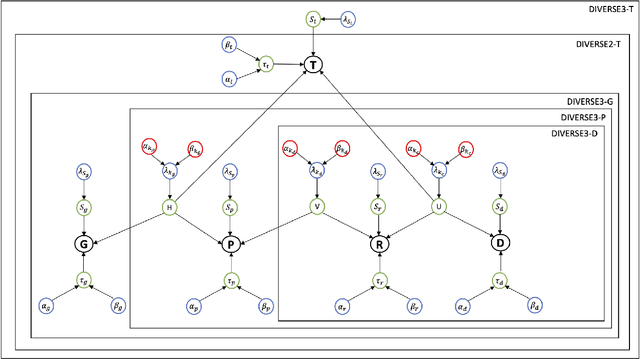
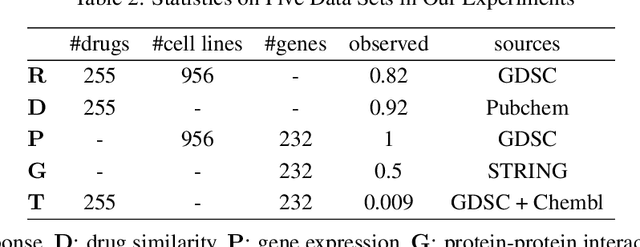
Detecting predictive biomarkers from multi-omics data is important for precision medicine, to improve diagnostics of complex diseases and for better treatments. This needs substantial experimental efforts that are made difficult by the heterogeneity of cell lines and huge cost. An effective solution is to build a computational model over the diverse omics data, including genomic, molecular, and environmental information. However, choosing informative and reliable data sources from among the different types of data is a challenging problem. We propose DIVERSE, a framework of Bayesian importance-weighted tri- and bi-matrix factorization(DIVERSE3 or DIVERSE2) to predict drug responses from data of cell lines, drugs, and gene interactions. DIVERSE integrates the data sources systematically, in a step-wise manner, examining the importance of each added data set in turn. More specifically, we sequentially integrate five different data sets, which have not all been combined in earlier bioinformatic methods for predicting drug responses. Empirical experiments show that DIVERSE clearly outperformed five other methods including three state-of-the-art approaches, under cross-validation, particularly in out-of-matrix prediction, which is closer to the setting of real use cases and more challenging than simpler in-matrix prediction. Additionally, case studies for discovering new drugs further confirmed the performance advantage of DIVERSE.
d3p -- A Python Package for Differentially-Private Probabilistic Programming
Mar 22, 2021
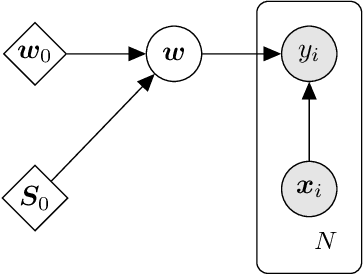
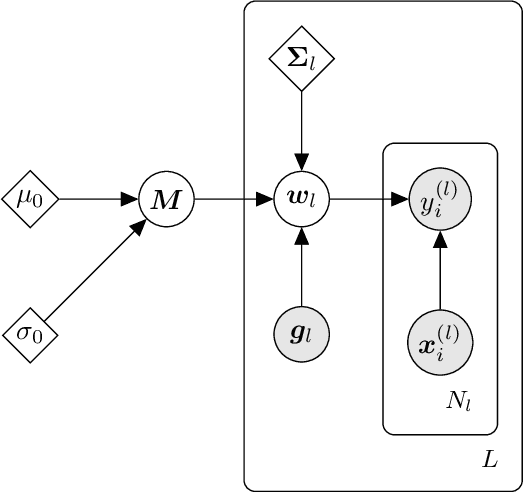

We present d3p, a software package designed to help fielding runtime efficient widely-applicable Bayesian inference under differential privacy guarantees. d3p achieves general applicability to a wide range of probabilistic modelling problems by implementing the differentially private variational inference algorithm, allowing users to fit any parametric probabilistic model with a differentiable density function. d3p adopts the probabilistic programming paradigm as a powerful way for the user to flexibly define such models. We demonstrate the use of our software on a hierarchical logistic regression example, showing the expressiveness of the modelling approach as well as the ease of running the parameter inference. We also perform an empirical evaluation of the runtime of the private inference on a complex model and find an $\sim$10 fold speed-up compared to an implementation using TensorFlow Privacy.
Decision Rule Elicitation for Domain Adaptation
Feb 23, 2021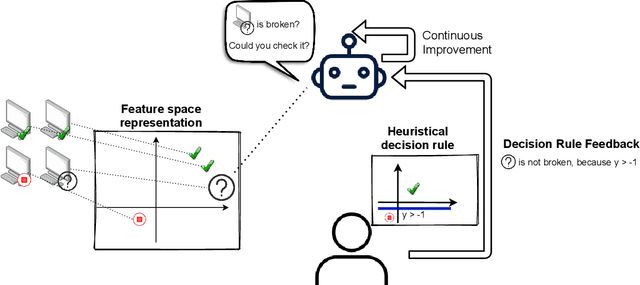
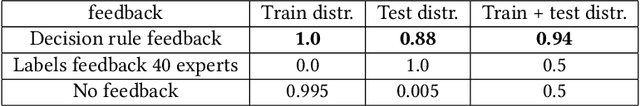
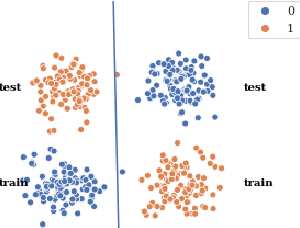
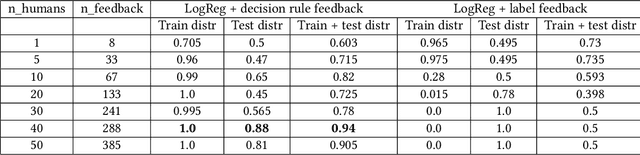
Human-in-the-loop machine learning is widely used in artificial intelligence (AI) to elicit labels for data points from experts or to provide feedback on how close the predicted results are to the target. This simplifies away all the details of the decision-making process of the expert. In this work, we allow the experts to additionally produce decision rules describing their decision-making; the rules are expected to be imperfect but to give additional information. In particular, the rules can extend to new distributions, and hence enable significantly improving performance for cases where the training and testing distributions differ, such as in domain adaptation. We apply the proposed method to lifelong learning and domain adaptation problems and discuss applications in other branches of AI, such as knowledge acquisition problems in expert systems. In simulated and real-user studies, we show that decision rule elicitation improves domain adaptation of the algorithm and helps to propagate expert's knowledge to the AI model.
Differentially Private Bayesian Inference for Generalized Linear Models
Nov 09, 2020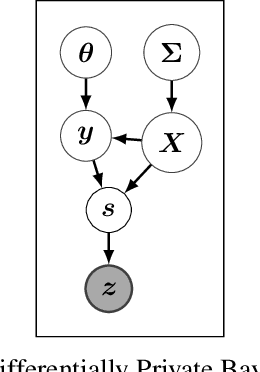
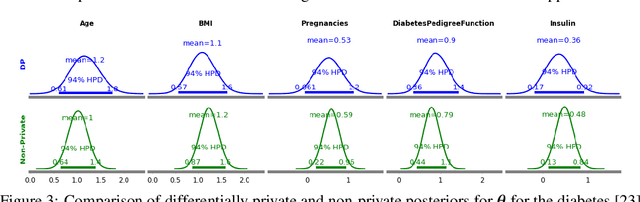
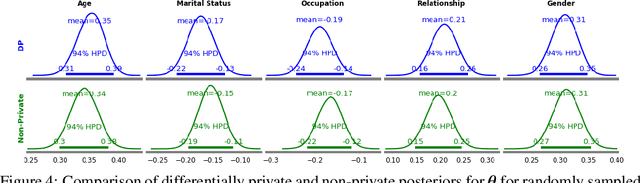
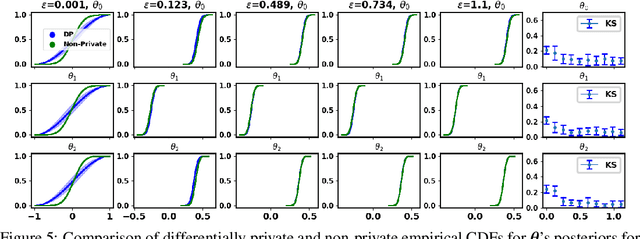
The framework of differential privacy (DP) upper bounds the information disclosure risk involved in using sensitive datasets for statistical analysis. A DP mechanism typically operates by adding carefully calibrated noise to the data release procedure. Generalized linear models (GLMs) are among the most widely used arms in data analyst's repertoire. In this work, with logistic and Poisson regression as running examples, we propose a generic noise-aware Bayesian framework to quantify the parameter uncertainty for a GLM at hand, given noisy sufficient statistics. We perform a tight privacy analysis and experimentally demonstrate that the posteriors obtained from our model, while adhering to strong privacy guarantees, are similar to the non-private posteriors.