 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeThe data heat island effect: quantifying the impact of AI data centers in a warming world
Mar 21, 2026The strong and continuous increase of AI-based services leads to the steady proliferation of AI data centres worldwide with the unavoidable escalation of their power consumption. It is unknown how this energy demand for computational purposes will impact the surrounding environment. Here, we focus our attention on the heat dissipation of AI hyperscalers. Taking advantage of land surface temperature measurements acquired by remote sensing platforms over the last decades, we are able to obtain a robust assessment of the temperature increase recorded in the areas surrounding AI data centres globally. We estimate that the land surface temperature increases by 2°C on average after the start of operations of an AI data centre, inducing local microclimate zones, which we call the data heat island effect. We assess the impact on the communities, quantifying that more than 340 million people could be affected by this temperature increase. Our results show that the data heat island effect could have a remarkable influence on communities and regional welfare in the future, hence becoming part of the conversation around environmentally sustainable AI worldwide.
CALF: Communication-Aware Learning Framework for Distributed Reinforcement Learning
Mar 13, 2026Distributed reinforcement learning policies face network delays, jitter, and packet loss when deployed across edge devices and cloud servers. Standard RL training assumes zero-latency interaction, causing severe performance degradation under realistic network conditions. We introduce CALF (Communication-Aware Learning Framework), which trains policies under realistic network models during simulation. Systematic experiments demonstrate that network-aware training substantially reduces deployment performance gaps compared to network-agnostic baselines. Distributed policy deployments across heterogeneous hardware validate that explicitly modelling communication constraints during training enables robust real-world execution. These findings establish network conditions as a major axis of sim-to-real transfer for Wi-Fi-like distributed deployments, complementing physics and visual domain randomisation.
Bridging Graph and State-Space Modeling for Intensive Care Unit Length of Stay Prediction
Aug 24, 2025Predicting a patient's length of stay (LOS) in the intensive care unit (ICU) is a critical task for hospital resource management, yet remains challenging due to the heterogeneous and irregularly sampled nature of electronic health records (EHRs). In this work, we propose S$^2$G-Net, a novel neural architecture that unifies state-space sequence modeling with multi-view Graph Neural Networks (GNNs) for ICU LOS prediction. The temporal path employs Mamba state-space models (SSMs) to capture patient trajectories, while the graph path leverages an optimized GraphGPS backbone, designed to integrate heterogeneous patient similarity graphs derived from diagnostic, administrative, and semantic features. Experiments on the large-scale MIMIC-IV cohort dataset show that S$^2$G-Net consistently outperforms sequence models (BiLSTM, Mamba, Transformer), graph models (classic GNNs, GraphGPS), and hybrid approaches across all primary metrics. Extensive ablation studies and interpretability analyses highlight the complementary contributions of each component of our architecture and underscore the importance of principled graph construction. These results demonstrate that S$^2$G-Net provides an effective and scalable solution for ICU LOS prediction with multi-modal clinical data.
Leveraging graph neural networks for supporting Automatic Triage of Patients
Mar 11, 2024Patient triage plays a crucial role in emergency departments, ensuring timely and appropriate care based on correctly evaluating the emergency grade of patient conditions. Triage methods are generally performed by human operator based on her own experience and information that are gathered from the patient management process. Thus, it is a process that can generate errors in emergency level associations. Recently, Traditional triage methods heavily rely on human decisions, which can be subjective and prone to errors. Recently, a growing interest has been focused on leveraging artificial intelligence (AI) to develop algorithms able to maximize information gathering and minimize errors in patient triage processing. We define and implement an AI based module to manage patients emergency code assignments in emergency departments. It uses emergency department historical data to train the medical decision process. Data containing relevant patient information, such as vital signs, symptoms, and medical history, are used to accurately classify patients into triage categories. Experimental results demonstrate that the proposed algorithm achieved high accuracy outperforming traditional triage methods. By using the proposed method we claim that healthcare professionals can predict severity index to guide patient management processing and resource allocation.
Improving embedding of graphs with missing data by soft manifolds
Nov 29, 2023



Embedding graphs in continous spaces is a key factor in designing and developing algorithms for automatic information extraction to be applied in diverse tasks (e.g., learning, inferring, predicting). The reliability of graph embeddings directly depends on how much the geometry of the continuous space matches the graph structure. Manifolds are mathematical structure that can enable to incorporate in their topological spaces the graph characteristics, and in particular nodes distances. State-of-the-art of manifold-based graph embedding algorithms take advantage of the assumption that the projection on a tangential space of each point in the manifold (corresponding to a node in the graph) would locally resemble a Euclidean space. Although this condition helps in achieving efficient analytical solutions to the embedding problem, it does not represent an adequate set-up to work with modern real life graphs, that are characterized by weighted connections across nodes often computed over sparse datasets with missing records. In this work, we introduce a new class of manifold, named soft manifold, that can solve this situation. In particular, soft manifolds are mathematical structures with spherical symmetry where the tangent spaces to each point are hypocycloids whose shape is defined according to the velocity of information propagation across the data points. Using soft manifolds for graph embedding, we can provide continuous spaces to pursue any task in data analysis over complex datasets. Experimental results on reconstruction tasks on synthetic and real datasets show how the proposed approach enable more accurate and reliable characterization of graphs in continuous spaces with respect to the state-of-the-art.
Interpretable Neural-Symbolic Concept Reasoning
Apr 27, 2023



Deep learning methods are highly accurate, yet their opaque decision process prevents them from earning full human trust. Concept-based models aim to address this issue by learning tasks based on a set of human-understandable concepts. However, state-of-the-art concept-based models rely on high-dimensional concept embedding representations which lack a clear semantic meaning, thus questioning the interpretability of their decision process. To overcome this limitation, we propose the Deep Concept Reasoner (DCR), the first interpretable concept-based model that builds upon concept embeddings. In DCR, neural networks do not make task predictions directly, but they build syntactic rule structures using concept embeddings. DCR then executes these rules on meaningful concept truth degrees to provide a final interpretable and semantically-consistent prediction in a differentiable manner. Our experiments show that DCR: (i) improves up to +25% w.r.t. state-of-the-art interpretable concept-based models on challenging benchmarks (ii) discovers meaningful logic rules matching known ground truths even in the absence of concept supervision during training, and (iii), facilitates the generation of counterfactual examples providing the learnt rules as guidance.
Survey on Leveraging Uncertainty Estimation Towards Trustworthy Deep Neural Networks: The Case of Reject Option and Post-training Processing
Apr 11, 2023



Although neural networks (especially deep neural networks) have achieved \textit{better-than-human} performance in many fields, their real-world deployment is still questionable due to the lack of awareness about the limitation in their knowledge. To incorporate such awareness in the machine learning model, prediction with reject option (also known as selective classification or classification with abstention) has been proposed in literature. In this paper, we present a systematic review of the prediction with the reject option in the context of various neural networks. To the best of our knowledge, this is the first study focusing on this aspect of neural networks. Moreover, we discuss different novel loss functions related to the reject option and post-training processing (if any) of network output for generating suitable measurements for knowledge awareness of the model. Finally, we address the application of the rejection option in reducing the prediction time for the real-time problems and present a comprehensive summary of the techniques related to the reject option in the context of extensive variety of neural networks. Our code is available on GitHub: \url{https://github.com/MehediHasanTutul/Reject_option}
On Over-Squashing in Message Passing Neural Networks: The Impact of Width, Depth, and Topology
Feb 06, 2023



Message Passing Neural Networks (MPNNs) are instances of Graph Neural Networks that leverage the graph to send messages over the edges. This inductive bias leads to a phenomenon known as over-squashing, where a node feature is insensitive to information contained at distant nodes. Despite recent methods introduced to mitigate this issue, an understanding of the causes for over-squashing and of possible solutions are lacking. In this theoretical work, we prove that: (i) Neural network width can mitigate over-squashing, but at the cost of making the whole network more sensitive; (ii) Conversely, depth cannot help mitigate over-squashing: increasing the number of layers leads to over-squashing being dominated by vanishing gradients; (iii) The graph topology plays the greatest role, since over-squashing occurs between nodes at high commute (access) time. Our analysis provides a unified framework to study different recent methods introduced to cope with over-squashing and serves as a justification for a class of methods that fall under `graph rewiring'.
Graph-Conditioned MLP for High-Dimensional Tabular Biomedical Data
Nov 11, 2022



Genome-wide studies leveraging recent high-throughput sequencing technologies collect high-dimensional data. However, they usually include small cohorts of patients, and the resulting tabular datasets suffer from the "curse of dimensionality". Training neural networks on such datasets is typically unstable, and the models overfit. One problem is that modern weight initialisation strategies make simplistic assumptions unsuitable for small-size datasets. We propose Graph-Conditioned MLP, a novel method to introduce priors on the parameters of an MLP. Instead of randomly initialising the first layer, we condition it directly on the training data. More specifically, we create a graph for each feature in the dataset (e.g., a gene), where each node represents a sample from the same dataset (e.g., a patient). We then use Graph Neural Networks (GNNs) to learn embeddings from these graphs and use the embeddings to initialise the MLP's parameters. Our approach opens the prospect of introducing additional biological knowledge when constructing the graphs. We present early results on 7 classification tasks from gene expression data and show that GC-MLP outperforms an MLP.
Graph Representation Learning on Tissue-Specific Multi-Omics
Jul 25, 2021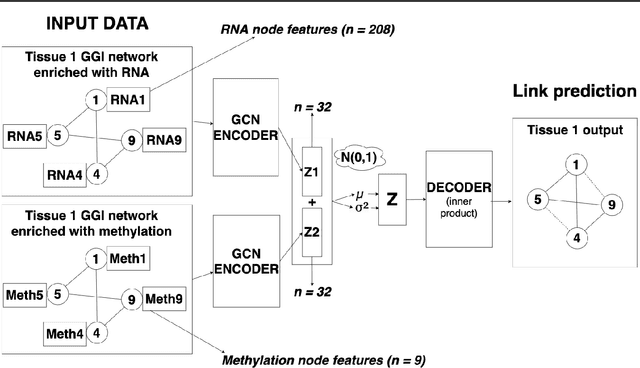
Combining different modalities of data from human tissues has been critical in advancing biomedical research and personalised medical care. In this study, we leverage a graph embedding model (i.e VGAE) to perform link prediction on tissue-specific Gene-Gene Interaction (GGI) networks. Through ablation experiments, we prove that the combination of multiple biological modalities (i.e multi-omics) leads to powerful embeddings and better link prediction performances. Our evaluation shows that the integration of gene methylation profiles and RNA-sequencing data significantly improves the link prediction performance. Overall, the combination of RNA-sequencing and gene methylation data leads to a link prediction accuracy of 71% on GGI networks. By harnessing graph representation learning on multi-omics data, our work brings novel insights to the current literature on multi-omics integration in bioinformatics.