 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAn LLM-Orchestrated Agent for Directional-Coupler Design with Self-Consistent Eigenmode and FDTD Validation
Jun 21, 2026We present a design agent which is a Large Language Model (LLM) that orchestrates, but does not perform, the numerical simulations to design a silicon-on-insulator (SOI) $2\times2$ directional coupler. We choose a symmetric phase-matched coupler where a lot of analytical results are available that help the design strategy. The LLM proposes candidate gap values (a geometrical dimension size) and judges convergence, while all physics is owned by deterministic solvers: a frequency-domain eigenmode solver estimates the coupling coefficient~$κ$ for the current design, and an independent Finite-Difference Time-Domain (FDTD) stage validates it. Both solvers operate on a common slab-projected two-dimensional (2D) effective-index reduction of the silicon film, so the design~$κ$ and the FDTD response are consistent by problem design; the residual between them is shown to be a single constant phase offset~$φ$, attributable to a fixed excess coupling length $L_{\mathrm{extra}}=\SI{2.837(11)}{\micro\meter}$ that we find invariant across a factor-of-two range in~$κ$. Folding this offset into a closed-loop length correction, the agent delivers a $50/50$ splitter whose FDTD-measured cross fraction is $0.498$ (target $0.500$), a residual of $0.0017$. Results are made self-consistent within the 2D effective-index model; and the LLM succeeds in delivering a suitable design over a number of attempts.
Vision-based Deep Learning Analysis of Unordered Biomedical Tabular Datasets via Optimal Spatial Cartography
Mar 24, 2026Tabular data are central to biomedical research, from liquid biopsy and bulk and single-cell transcriptomics to electronic health records and phenotypic profiling. Unlike images or sequences, however, tabular datasets lack intrinsic spatial organization: features are treated as unordered dimensions, and their relationships must be inferred implicitly by the model. This limits the ability of vision architectures to exploit local structure and higher-order feature interactions in non-spatial biomedical data. Here we introduce Dynamic Feature Mapping (Dynomap), an end-to-end deep learning framework that learns a task-optimized spatial topology of features directly from data. Dynomap jointly optimizes feature placement and prediction through a fully differentiable rendering mechanism, without relying on heuristics, predefined groupings, or external priors. By transforming high-dimensional tabular vectors into learned feature maps, Dynomap enables vision-based models to operate effectively on unordered biomedical inputs. Across multiple clinical and biological datasets, Dynomap consistently outperformed classical machine learning, modern deep tabular models, and existing vector-to-image approaches. In liquid biopsy data, Dynomap organized clinically relevant gene signatures into coherent spatial patterns and improved multiclass cancer subtype prediction accuracy by up to 18%. In a Parkinson disease voice dataset, it clustered disease-associated acoustic descriptors and improved accuracy by up to 8%. Similar gains and interpretable feature organization were observed in additional biomedical datasets. These results establish Dynomap as a general strategy for bridging tabular and vision-based deep learning and for uncovering structured, clinically relevant patterns in high-dimensional biomedical data.
Redefining the Down-Sampling Scheme of U-Net for Precision Biomedical Image Segmentation
Feb 23, 2026U-Net architectures have been instrumental in advancing biomedical image segmentation (BIS) but often struggle with capturing long-range information. One reason is the conventional down-sampling techniques that prioritize computational efficiency at the expense of information retention. This paper introduces a simple but effective strategy, we call it Stair Pooling, which moderates the pace of down-sampling and reduces information loss by leveraging a sequence of concatenated small and narrow pooling operations in varied orientations. Specifically, our method modifies the reduction in dimensionality within each 2D pooling step from $\frac{1}{4}$ to $\frac{1}{2}$. This approach can also be adapted for 3D pooling to preserve even more information. Such preservation aids the U-Net in more effectively reconstructing spatial details during the up-sampling phase, thereby enhancing its ability to capture long-range information and improving segmentation accuracy. Extensive experiments on three BIS benchmarks demonstrate that the proposed Stair Pooling can increase both 2D and 3D U-Net performance by an average of 3.8\% in Dice scores. Moreover, we leverage the transfer entropy to select the optimal down-sampling paths and quantitatively show how the proposed Stair Pooling reduces the information loss.
Uncovering spatial tissue domains and cell types in spatial omics through cross-scale profiling of cellular and genomic interactions
Feb 13, 2026Cellular identity and function are linked to both their intrinsic genomic makeup and extrinsic spatial context within the tissue microenvironment. Spatial transcriptomics (ST) offers an unprecedented opportunity to study this, providing in situ gene expression profiles at single-cell resolution and illuminating the spatial and functional organization of cells within tissues. However, a significant hurdle remains: ST data is inherently noisy, large, and structurally complex. This complexity makes it intractable for existing computational methods to effectively capture the interplay between spatial interactions and intrinsic genomic relationships, thus limiting our ability to discern critical biological patterns. Here, we present CellScape, a deep learning framework designed to overcome these limitations for high-performance ST data analysis and pattern discovery. CellScape jointly models cellular interactions in tissue space and genomic relationships among cells, producing comprehensive representations that seamlessly integrate spatial signals with underlying gene regulatory mechanisms. This technique uncovers biologically informative patterns that improve spatial domain segmentation and supports comprehensive spatial cellular analyses across diverse transcriptomics datasets, offering an accurate and versatile framework for deep analysis and interpretation of ST data.w
Deep-and-Wide Learning: Enhancing Data-Driven Inference via Synergistic Learning of Inter- and Intra-Data Representations
Jan 28, 2025



Advancements in deep learning are revolutionizing science and engineering. The immense success of deep learning is largely due to its ability to extract essential high-dimensional (HD) features from input data and make inference decisions based on this information. However, current deep neural network (DNN) models face several challenges, such as the requirements of extensive amounts of data and computational resources. Here, we introduce a new learning scheme, referred to as deep-and-wide learning (DWL), to systematically capture features not only within individual input data (intra-data features) but also across the data (inter-data features). Furthermore, we propose a dual-interactive-channel network (D-Net) to realize the DWL, which leverages our Bayesian formulation of low-dimensional (LD) inter-data feature extraction and its synergistic interaction with the conventional HD representation of the dataset, for substantially enhanced computational efficiency and inference. The proposed technique has been applied to data across various disciplines for both classification and regression tasks. Our results demonstrate that DWL surpasses state-of-the-art DNNs in accuracy by a substantial margin with limited training data and improves the computational efficiency by order(s) of magnitude. The proposed DWL strategy dramatically alters the data-driven learning techniques, including emerging large foundation models, and sheds significant insights into the evolving field of AI.
Discovering distinctive elements of biomedical datasets for high-performance exploration
Oct 07, 2024



The human brain represents an object by small elements and distinguishes two objects based on the difference in elements. Discovering the distinctive elements of high-dimensional datasets is therefore critical in numerous perception-driven biomedical and clinical studies. However, currently there is no available method for reliable extraction of distinctive elements of high-dimensional biomedical and clinical datasets. Here we present an unsupervised deep learning technique namely distinctive element analysis (DEA), which extracts the distinctive data elements using high-dimensional correlative information of the datasets. DEA at first computes a large number of distinctive parts of the data, then filters and condenses the parts into DEA elements by employing a unique kernel-driven triple-optimization network. DEA has been found to improve the accuracy by up to 45% in comparison to the traditional techniques in applications such as disease detection from medical images, gene ranking and cell recognition from single cell RNA sequence (scRNA-seq) datasets. Moreover, DEA allows user-guided manipulation of the intermediate calculation process and thus offers intermediate results with better interpretability.
A Hybrid Deep Feature-Based Deformable Image Registration Method for Pathological Images
Aug 17, 2022
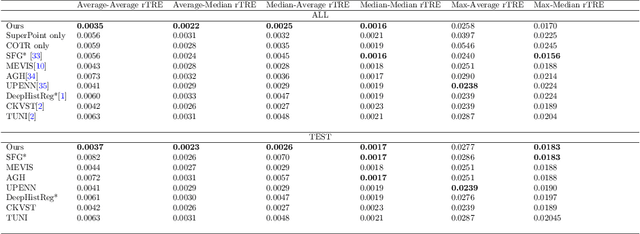
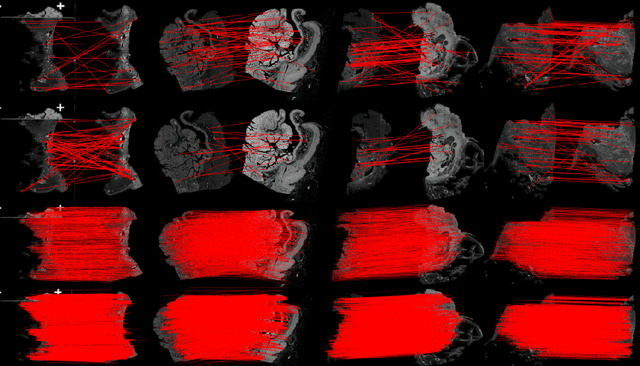
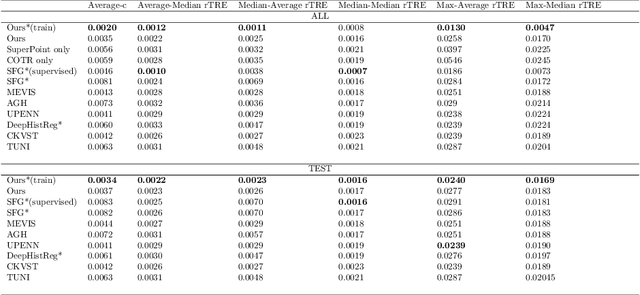
Pathologists need to combine information from differently stained pathological slices to obtain accurate diagnostic results. Deformable image registration is a necessary technique for fusing multi-modal pathological slices. This paper proposes a hybrid deep feature-based deformable image registration framework for stained pathological samples. We first extract dense feature points and perform points matching by two deep learning feature networks. Then, to further reduce false matches, an outlier detection method combining the isolation forest statistical model and the local affine correction model is proposed. Finally, the interpolation method generates the DVF for pathology image registration based on the above matching points. We evaluate our method on the dataset of the Non-rigid Histology Image Registration (ANHIR) challenge, which is co-organized with the IEEE ISBI 2019 conference. Our technique outperforms the traditional approaches by 17% with the Average-Average registration target error (rTRE) reaching 0.0034. The proposed method achieved state-of-the-art performance and ranking it 1 in evaluating the test dataset. The proposed hybrid deep feature-based registration method can potentially become a reliable method for pathology image registration.
Learning Treatment Plan Representations for Content Based Image Retrieval
Jun 06, 2022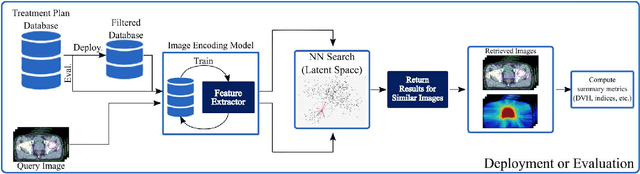

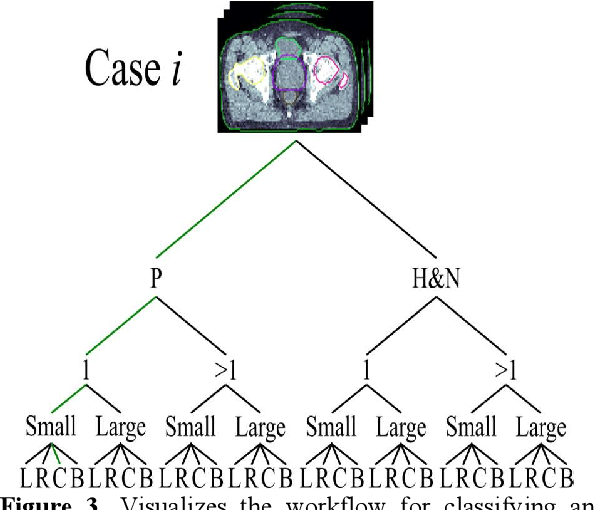
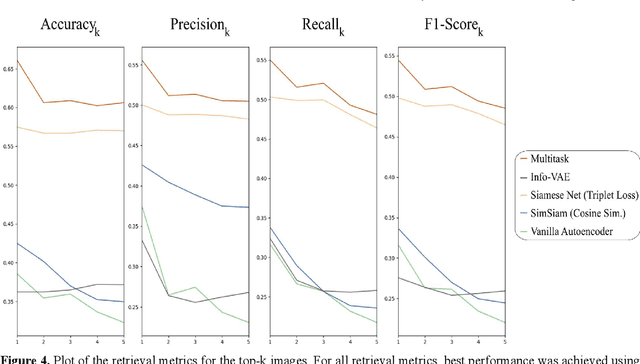
Objective: Knowledge based planning (KBP) typically involves training an end-to-end deep learning model to predict dose distributions. However, training end-to-end KBP methods may be associated with practical limitations due to the limited size of medical datasets that are often used. To address these limitations, we propose a content based image retrieval (CBIR) method for retrieving dose distributions of previously planned patients based on anatomical similarity. Approach: Our proposed CBIR method trains a representation model that produces latent space embeddings of a patient's anatomical information. The latent space embeddings of new patients are then compared against those of previous patients in a database for image retrieval of dose distributions. Summary metrics (e.g. dose-volume histogram, conformity index, homogeneity index, etc.) are computed and can then be utilized in subsequent automated planning. All source code for this project is available on github. Main Results: The retrieval performance of various CBIR methods is evaluated on a dataset consisting of both publicly available plans and clinical plans from our institution. This study compares various encoding methods, ranging from simple autoencoders to more recent Siamese networks like SimSiam, and the best performance was observed for the multitask Siamese network. Significance: Applying CBIR to inform subsequent treatment planning potentially addresses many limitations associated with end-to-end KBP. Our current results demonstrate that excellent image retrieval performance can be obtained through slight changes to previously developed Siamese networks. We hope to integrate CBIR into automated planning workflow in future works, potentially through methods like the MetaPlanner framework.
Image Classification using Graph Neural Network and Multiscale Wavelet Superpixels
Jan 29, 2022



Prior studies using graph neural networks (GNNs) for image classification have focused on graphs generated from a regular grid of pixels or similar-sized superpixels. In the latter, a single target number of superpixels is defined for an entire dataset irrespective of differences across images and their intrinsic multiscale structure. On the contrary, this study investigates image classification using graphs generated from an image-specific number of multiscale superpixels. We propose WaveMesh, a new wavelet-based superpixeling algorithm, where the number and sizes of superpixels in an image are systematically computed based on its content. WaveMesh superpixel graphs are structurally different from similar-sized superpixel graphs. We use SplineCNN, a state-of-the-art network for image graph classification, to compare WaveMesh and similar-sized superpixels. Using SplineCNN, we perform extensive experiments on three benchmark datasets under three local-pooling settings: 1) no pooling, 2) GraclusPool, and 3) WavePool, a novel spatially heterogeneous pooling scheme tailored to WaveMesh superpixels. Our experiments demonstrate that SplineCNN learns from multiscale WaveMesh superpixels on-par with similar-sized superpixels. In all WaveMesh experiments, GraclusPool performs poorer than no pooling / WavePool, indicating that poor choice of pooling can result in inferior performance while learning from multiscale superpixels.
Self-supervised Feature Learning via Exploiting Multi-modal Data for Retinal Disease Diagnosis
Jul 21, 2020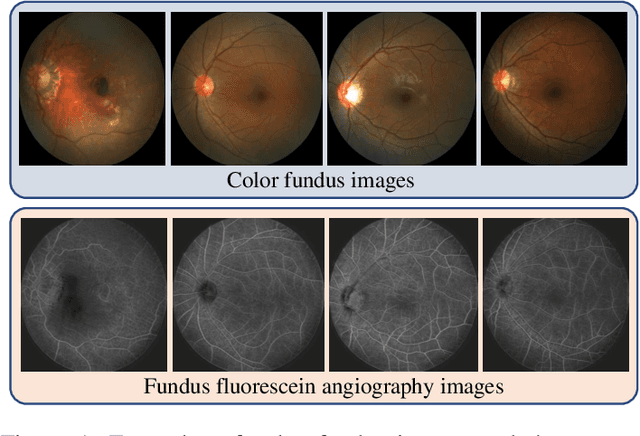
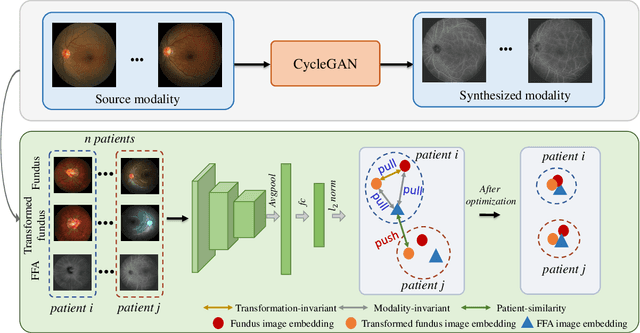
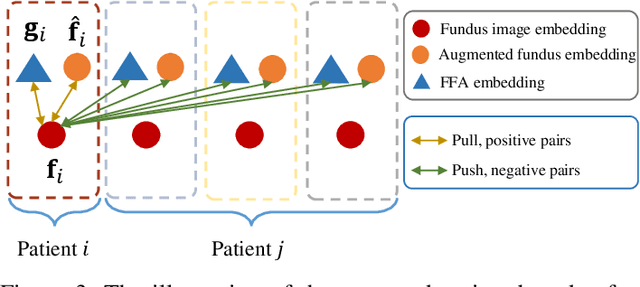
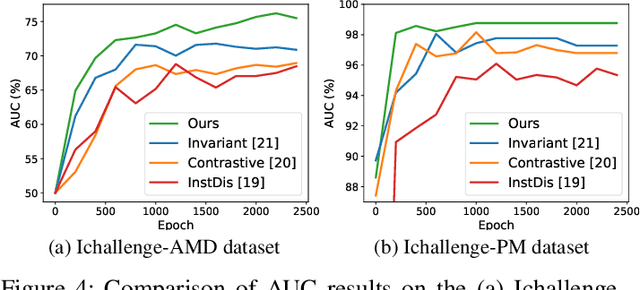
The automatic diagnosis of various retinal diseases from fundus images is important to support clinical decision-making. However, developing such automatic solutions is challenging due to the requirement of a large amount of human-annotated data. Recently, unsupervised/self-supervised feature learning techniques receive a lot of attention, as they do not need massive annotations. Most of the current self-supervised methods are analyzed with single imaging modality and there is no method currently utilize multi-modal images for better results. Considering that the diagnostics of various vitreoretinal diseases can greatly benefit from another imaging modality, e.g., FFA, this paper presents a novel self-supervised feature learning method by effectively exploiting multi-modal data for retinal disease diagnosis. To achieve this, we first synthesize the corresponding FFA modality and then formulate a patient feature-based softmax embedding objective. Our objective learns both modality-invariant features and patient-similarity features. Through this mechanism, the neural network captures the semantically shared information across different modalities and the apparent visual similarity between patients. We evaluate our method on two public benchmark datasets for retinal disease diagnosis. The experimental results demonstrate that our method clearly outperforms other self-supervised feature learning methods and is comparable to the supervised baseline.