 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMeta Flow Matching: Integrating Vector Fields on the Wasserstein Manifold
Aug 26, 2024



Numerous biological and physical processes can be modeled as systems of interacting entities evolving continuously over time, e.g. the dynamics of communicating cells or physical particles. Learning the dynamics of such systems is essential for predicting the temporal evolution of populations across novel samples and unseen environments. Flow-based models allow for learning these dynamics at the population level - they model the evolution of the entire distribution of samples. However, current flow-based models are limited to a single initial population and a set of predefined conditions which describe different dynamics. We argue that multiple processes in natural sciences have to be represented as vector fields on the Wasserstein manifold of probability densities. That is, the change of the population at any moment in time depends on the population itself due to the interactions between samples. In particular, this is crucial for personalized medicine where the development of diseases and their respective treatment response depends on the microenvironment of cells specific to each patient. We propose Meta Flow Matching (MFM), a practical approach to integrating along these vector fields on the Wasserstein manifold by amortizing the flow model over the initial populations. Namely, we embed the population of samples using a Graph Neural Network (GNN) and use these embeddings to train a Flow Matching model. This gives MFM the ability to generalize over the initial distributions unlike previously proposed methods. We demonstrate the ability of MFM to improve prediction of individual treatment responses on a large scale multi-patient single-cell drug screen dataset.
Splicing Up Your Predictions with RNA Contrastive Learning
Oct 17, 2023



In the face of rapidly accumulating genomic data, our understanding of the RNA regulatory code remains incomplete. Recent self-supervised methods in other domains have demonstrated the ability to learn rules underlying the data-generating process such as sentence structure in language. Inspired by this, we extend contrastive learning techniques to genomic data by utilizing functional similarities between sequences generated through alternative splicing and gene duplication. Our novel dataset and contrastive objective enable the learning of generalized RNA isoform representations. We validate their utility on downstream tasks such as RNA half-life and mean ribosome load prediction. Our pre-training strategy yields competitive results using linear probing on both tasks, along with up to a two-fold increase in Pearson correlation in low-data conditions. Importantly, our exploration of the learned latent space reveals that our contrastive objective yields semantically meaningful representations, underscoring its potential as a valuable initialization technique for RNA property prediction.
DynGFN: Bayesian Dynamic Causal Discovery using Generative Flow Networks
Feb 08, 2023



Learning the causal structure of observable variables is a central focus for scientific discovery. Bayesian causal discovery methods tackle this problem by learning a posterior over the set of admissible graphs given our priors and observations. Existing methods primarily consider observations from static systems and assume the underlying causal structure takes the form of a directed acyclic graph (DAG). In settings with dynamic feedback mechanisms that regulate the trajectories of individual variables, this acyclicity assumption fails unless we account for time. We focus on learning Bayesian posteriors over cyclic graphs and treat causal discovery as a problem of sparse identification of a dynamical system. This imposes a natural temporal causal order between variables and captures cyclic feedback loops through time. Under this lens, we propose a new framework for Bayesian causal discovery for dynamical systems and present a novel generative flow network architecture (DynGFN) tailored for this task. Our results indicate that DynGFN learns posteriors that better encapsulate the distributions over admissible cyclic causal structures compared to counterpart state-of-the-art approaches.
Generating and designing DNA with deep generative models
Dec 17, 2017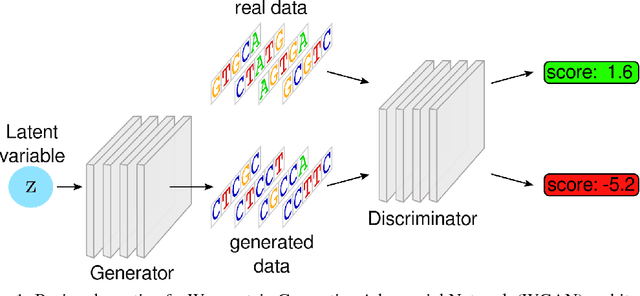
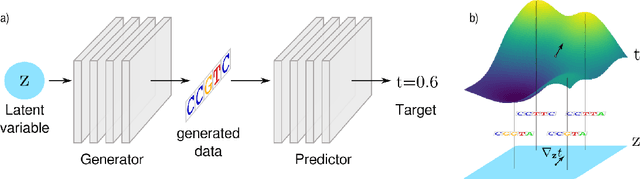
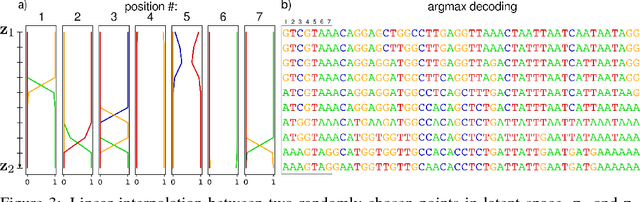
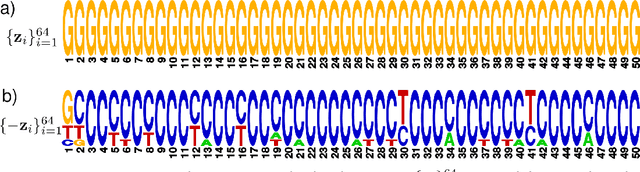
We propose generative neural network methods to generate DNA sequences and tune them to have desired properties. We present three approaches: creating synthetic DNA sequences using a generative adversarial network; a DNA-based variant of the activation maximization ("deep dream") design method; and a joint procedure which combines these two approaches together. We show that these tools capture important structures of the data and, when applied to designing probes for protein binding microarrays, allow us to generate new sequences whose properties are estimated to be superior to those found in the training data. We believe that these results open the door for applying deep generative models to advance genomics research.