 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRe-identification from histopathology images
Mar 19, 2024



In numerous studies, deep learning algorithms have proven their potential for the analysis of histopathology images, for example, for revealing the subtypes of tumors or the primary origin of metastases. These models require large datasets for training, which must be anonymized to prevent possible patient identity leaks. This study demonstrates that even relatively simple deep learning algorithms can re-identify patients in large histopathology datasets with substantial accuracy. We evaluated our algorithms on two TCIA datasets including lung squamous cell carcinoma (LSCC) and lung adenocarcinoma (LUAD). We also demonstrate the algorithm's performance on an in-house dataset of meningioma tissue. We predicted the source patient of a slide with F1 scores of 50.16 % and 52.30 % on the LSCC and LUAD datasets, respectively, and with 62.31 % on our meningioma dataset. Based on our findings, we formulated a risk assessment scheme to estimate the risk to the patient's privacy prior to publication.
Rethinking U-net Skip Connections for Biomedical Image Segmentation
Feb 13, 2024The U-net architecture has significantly impacted deep learning-based segmentation of medical images. Through the integration of long-range skip connections, it facilitated the preservation of high-resolution features. Out-of-distribution data can, however, substantially impede the performance of neural networks. Previous works showed that the trained network layers differ in their susceptibility to this domain shift, e.g., shallow layers are more affected than deeper layers. In this work, we investigate the implications of this observation of layer sensitivity to domain shifts of U-net-style segmentation networks. By copying features of shallow layers to corresponding decoder blocks, these bear the risk of re-introducing domain-specific information. We used a synthetic dataset to model different levels of data distribution shifts and evaluated the impact on downstream segmentation performance. We quantified the inherent domain susceptibility of each network layer, using the Hellinger distance. These experiments confirmed the higher domain susceptibility of earlier network layers. When gradually removing skip connections, a decrease in domain susceptibility of deeper layers could be observed. For downstream segmentation performance, the original U-net outperformed the variant without any skip connections. The best performance, however, was achieved when removing the uppermost skip connection - not only in the presence of domain shifts but also for in-domain test data. We validated our results on three clinical datasets - two histopathology datasets and one magnetic resonance dataset - with performance increases of up to 10% in-domain and 13% cross-domain when removing the uppermost skip connection.
Deep Learning model predicts the c-Kit-11 mutational status of canine cutaneous mast cell tumors by HE stained histological slides
Jan 02, 2024



Numerous prognostic factors are currently assessed histopathologically in biopsies of canine mast cell tumors to evaluate clinical behavior. In addition, PCR analysis of the c-Kit exon 11 mutational status is often performed to evaluate the potential success of a tyrosine kinase inhibitor therapy. This project aimed at training deep learning models (DLMs) to identify the c-Kit-11 mutational status of MCTs solely based on morphology without additional molecular analysis. HE slides of 195 mutated and 173 non-mutated tumors were stained consecutively in two different laboratories and scanned with three different slide scanners. This resulted in six different datasets (stain-scanner variations) of whole slide images. DLMs were trained with single and mixed datasets and their performances was assessed under scanner and staining domain shifts. The DLMs correctly classified HE slides according to their c-Kit 11 mutation status in, on average, 87% of cases for the best-suited stain-scanner variant. A relevant performance drop could be observed when the stain-scanner combination of the training and test dataset differed. Multi-variant datasets improved the average accuracy but did not reach the maximum accuracy of algorithms trained and tested on the same stain-scanner variant. In summary, DLM-assisted morphological examination of MCTs can predict c-Kit-exon 11 mutational status of MCTs with high accuracy. However, the recognition performance is impeded by a change of scanner or staining protocol. Larger data sets with higher numbers of scans originating from different laboratories and scanners may lead to more robust DLMs to identify c-Kit mutations in HE slides.
Automated Volume Corrected Mitotic Index Calculation Through Annotation-Free Deep Learning using Immunohistochemistry as Reference Standard
Nov 15, 2023



The volume-corrected mitotic index (M/V-Index) was shown to provide prognostic value in invasive breast carcinomas. However, despite its prognostic significance, it is not established as the standard method for assessing aggressive biological behaviour, due to the high additional workload associated with determining the epithelial proportion. In this work, we show that using a deep learning pipeline solely trained with an annotation-free, immunohistochemistry-based approach, provides accurate estimations of epithelial segmentation in canine breast carcinomas. We compare our automatic framework with the manually annotated M/V-Index in a study with three board-certified pathologists. Our results indicate that the deep learning-based pipeline shows expert-level performance, while providing time efficiency and reproducibility.
Few Shot Learning for the Classification of Confocal Laser Endomicroscopy Images of Head and Neck Tumors
Nov 13, 2023


The surgical removal of head and neck tumors requires safe margins, which are usually confirmed intraoperatively by means of frozen sections. This method is, in itself, an oversampling procedure, which has a relatively low sensitivity compared to the definitive tissue analysis on paraffin-embedded sections. Confocal laser endomicroscopy (CLE) is an in-vivo imaging technique that has shown its potential in the live optical biopsy of tissue. An automated analysis of this notoriously difficult to interpret modality would help surgeons. However, the images of CLE show a wide variability of patterns, caused both by individual factors but also, and most strongly, by the anatomical structures of the imaged tissue, making it a challenging pattern recognition task. In this work, we evaluate four popular few shot learning (FSL) methods towards their capability of generalizing to unseen anatomical domains in CLE images. We evaluate this on images of sinunasal tumors (SNT) from five patients and on images of the vocal folds (VF) from 11 patients using a cross-validation scheme. The best respective approach reached a median accuracy of 79.6% on the rather homogeneous VF dataset, but only of 61.6% for the highly diverse SNT dataset. Our results indicate that FSL on CLE images is viable, but strongly affected by the number of patients, as well as the diversity of anatomical patterns.
Domain generalization across tumor types, laboratories, and species -- insights from the 2022 edition of the Mitosis Domain Generalization Challenge
Sep 27, 2023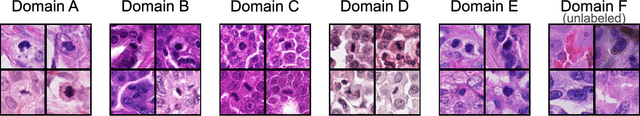
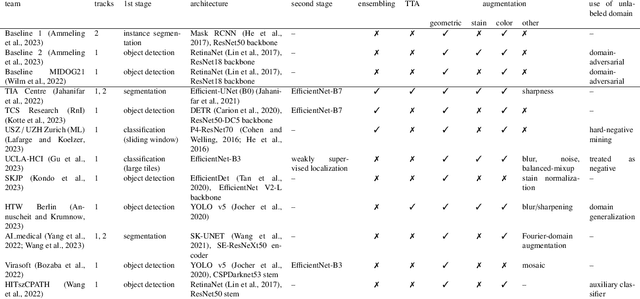
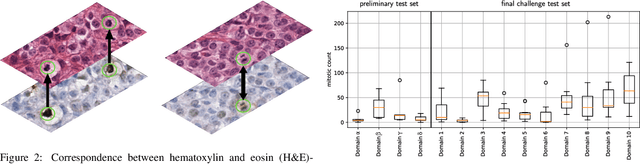
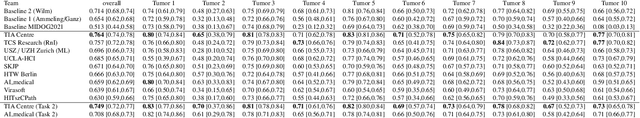
Recognition of mitotic figures in histologic tumor specimens is highly relevant to patient outcome assessment. This task is challenging for algorithms and human experts alike, with deterioration of algorithmic performance under shifts in image representations. Considerable covariate shifts occur when assessment is performed on different tumor types, images are acquired using different digitization devices, or specimens are produced in different laboratories. This observation motivated the inception of the 2022 challenge on MItosis Domain Generalization (MIDOG 2022). The challenge provided annotated histologic tumor images from six different domains and evaluated the algorithmic approaches for mitotic figure detection provided by nine challenge participants on ten independent domains. Ground truth for mitotic figure detection was established in two ways: a three-expert consensus and an independent, immunohistochemistry-assisted set of labels. This work represents an overview of the challenge tasks, the algorithmic strategies employed by the participants, and potential factors contributing to their success. With an $F_1$ score of 0.764 for the top-performing team, we summarize that domain generalization across various tumor domains is possible with today's deep learning-based recognition pipelines. When assessed against the immunohistochemistry-assisted reference standard, all methods resulted in reduced recall scores, but with only minor changes in the order of participants in the ranking.
Nuclear Morphometry using a Deep Learning-based Algorithm has Prognostic Relevance for Canine Cutaneous Mast Cell Tumors
Sep 26, 2023
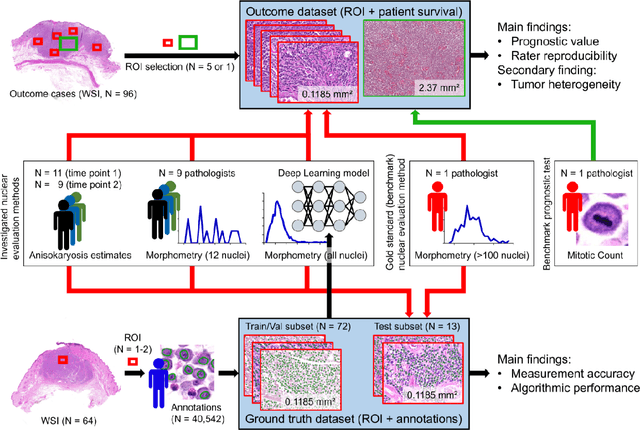


Variation in nuclear size and shape is an important criterion of malignancy for many tumor types; however, categorical estimates by pathologists have poor reproducibility. Measurements of nuclear characteristics (morphometry) can improve reproducibility, but manual methods are time consuming. In this study, we evaluated fully automated morphometry using a deep learning-based algorithm in 96 canine cutaneous mast cell tumors with information on patient survival. Algorithmic morphometry was compared with karyomegaly estimates by 11 pathologists, manual nuclear morphometry of 12 cells by 9 pathologists, and the mitotic count as a benchmark. The prognostic value of automated morphometry was high with an area under the ROC curve regarding the tumor-specific survival of 0.943 (95% CI: 0.889 - 0.996) for the standard deviation (SD) of nuclear area, which was higher than manual morphometry of all pathologists combined (0.868, 95% CI: 0.737 - 0.991) and the mitotic count (0.885, 95% CI: 0.765 - 1.00). At the proposed thresholds, the hazard ratio for algorithmic morphometry (SD of nuclear area $\geq 9.0 \mu m^2$) was 18.3 (95% CI: 5.0 - 67.1), for manual morphometry (SD of nuclear area $\geq 10.9 \mu m^2$) 9.0 (95% CI: 6.0 - 13.4), for karyomegaly estimates 7.6 (95% CI: 5.7 - 10.1), and for the mitotic count 30.5 (95% CI: 7.8 - 118.0). Inter-rater reproducibility for karyomegaly estimates was fair ($\kappa$ = 0.226) with highly variable sensitivity/specificity values for the individual pathologists. Reproducibility for manual morphometry (SD of nuclear area) was good (ICC = 0.654). This study supports the use of algorithmic morphometry as a prognostic test to overcome the limitations of estimates and manual measurements.
Adaptive Region Selection for Active Learning in Whole Slide Image Semantic Segmentation
Jul 14, 2023



The process of annotating histological gigapixel-sized whole slide images (WSIs) at the pixel level for the purpose of training a supervised segmentation model is time-consuming. Region-based active learning (AL) involves training the model on a limited number of annotated image regions instead of requesting annotations of the entire images. These annotation regions are iteratively selected, with the goal of optimizing model performance while minimizing the annotated area. The standard method for region selection evaluates the informativeness of all square regions of a specified size and then selects a specific quantity of the most informative regions. We find that the efficiency of this method highly depends on the choice of AL step size (i.e., the combination of region size and the number of selected regions per WSI), and a suboptimal AL step size can result in redundant annotation requests or inflated computation costs. This paper introduces a novel technique for selecting annotation regions adaptively, mitigating the reliance on this AL hyperparameter. Specifically, we dynamically determine each region by first identifying an informative area and then detecting its optimal bounding box, as opposed to selecting regions of a uniform predefined shape and size as in the standard method. We evaluate our method using the task of breast cancer metastases segmentation on the public CAMELYON16 dataset and show that it consistently achieves higher sampling efficiency than the standard method across various AL step sizes. With only 2.6\% of tissue area annotated, we achieve full annotation performance and thereby substantially reduce the costs of annotating a WSI dataset. The source code is available at https://github.com/DeepMicroscopy/AdaptiveRegionSelection.
Wuerstchen: Efficient Pretraining of Text-to-Image Models
Jun 01, 2023We introduce Wuerstchen, a novel technique for text-to-image synthesis that unites competitive performance with unprecedented cost-effectiveness and ease of training on constrained hardware. Building on recent advancements in machine learning, our approach, which utilizes latent diffusion strategies at strong latent image compression rates, significantly reduces the computational burden, typically associated with state-of-the-art models, while preserving, if not enhancing, the quality of generated images. Wuerstchen achieves notable speed improvements at inference time, thereby rendering real-time applications more viable. One of the key advantages of our method lies in its modest training requirements of only 9,200 GPU hours, slashing the usual costs significantly without compromising the end performance. In a comparison against the state-of-the-art, we found the approach to yield strong competitiveness. This paper opens the door to a new line of research that prioritizes both performance and computational accessibility, hence democratizing the use of sophisticated AI technologies. Through Wuerstchen, we demonstrate a compelling stride forward in the realm of text-to-image synthesis, offering an innovative path to explore in future research.
Why is the winner the best?
Mar 30, 2023



International benchmarking competitions have become fundamental for the comparative performance assessment of image analysis methods. However, little attention has been given to investigating what can be learnt from these competitions. Do they really generate scientific progress? What are common and successful participation strategies? What makes a solution superior to a competing method? To address this gap in the literature, we performed a multi-center study with all 80 competitions that were conducted in the scope of IEEE ISBI 2021 and MICCAI 2021. Statistical analyses performed based on comprehensive descriptions of the submitted algorithms linked to their rank as well as the underlying participation strategies revealed common characteristics of winning solutions. These typically include the use of multi-task learning (63%) and/or multi-stage pipelines (61%), and a focus on augmentation (100%), image preprocessing (97%), data curation (79%), and postprocessing (66%). The "typical" lead of a winning team is a computer scientist with a doctoral degree, five years of experience in biomedical image analysis, and four years of experience in deep learning. Two core general development strategies stood out for highly-ranked teams: the reflection of the metrics in the method design and the focus on analyzing and handling failure cases. According to the organizers, 43% of the winning algorithms exceeded the state of the art but only 11% completely solved the respective domain problem. The insights of our study could help researchers (1) improve algorithm development strategies when approaching new problems, and (2) focus on open research questions revealed by this work.