 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePyHealth: A Python Library for Health Predictive Models
Jan 11, 2021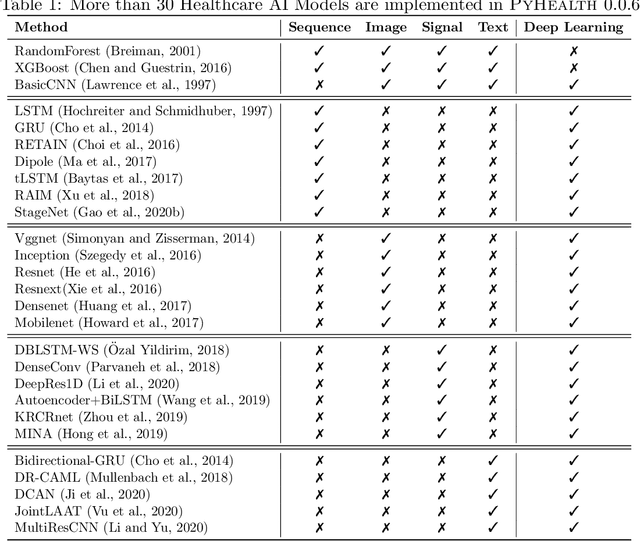
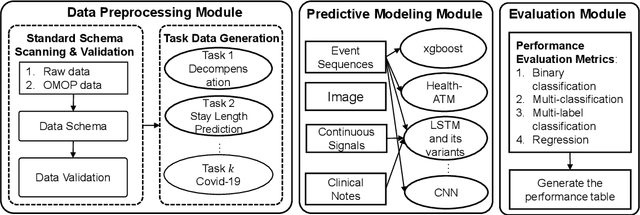
Despite the explosion of interest in healthcare AI research, the reproducibility and benchmarking of those research works are often limited due to the lack of standard benchmark datasets and diverse evaluation metrics. To address this reproducibility challenge, we develop PyHealth, an open-source Python toolbox for developing various predictive models on healthcare data. PyHealth consists of data preprocessing module, predictive modeling module, and evaluation module. The target users of PyHealth are both computer science researchers and healthcare data scientists. With PyHealth, they can conduct complex machine learning pipelines on healthcare datasets with fewer than ten lines of code. The data preprocessing module enables the transformation of complex healthcare datasets such as longitudinal electronic health records, medical images, continuous signals (e.g., electrocardiogram), and clinical notes into machine learning friendly formats. The predictive modeling module provides more than 30 machine learning models, including established ensemble trees and deep neural network-based approaches, via a unified but extendable API designed for both researchers and practitioners. The evaluation module provides various evaluation strategies (e.g., cross-validation and train-validation-test split) and predictive model metrics. With robustness and scalability in mind, best practices such as unit testing, continuous integration, code coverage, and interactive examples are introduced in the library's development. PyHealth can be installed through the Python Package Index (PyPI) or https://github.com/yzhao062/PyHealth .
Fast Graph Attention Networks Using Effective Resistance Based Graph Sparsification
Jun 17, 2020
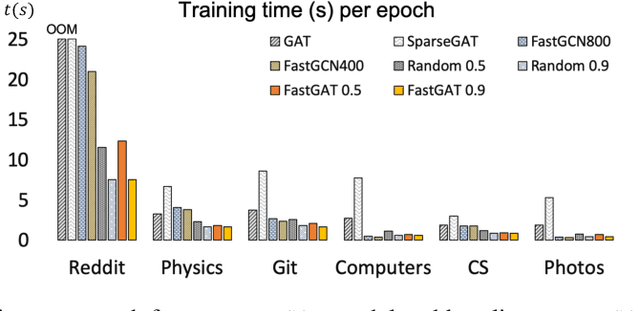
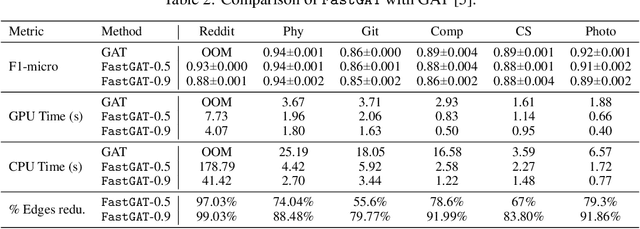
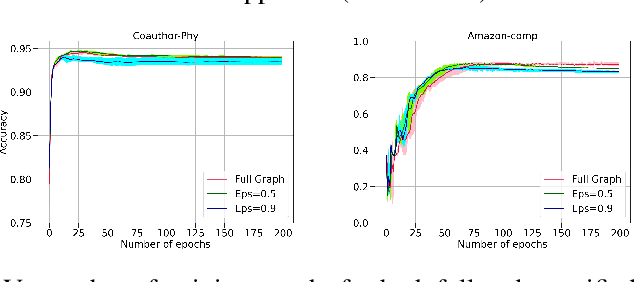
The attention mechanism has demonstrated superior performance for inference over nodes in graph neural networks (GNNs), however, they result in a high computational burden during both training and inference. We propose FastGAT, a method to make attention based GNNs lightweight by using spectral sparsification to generate an optimal pruning of the input graph. This results in a per-epoch time that is almost linear in the number of graph nodes as opposed to quadratic. Further, we provide a re-formulation of a specific attention based GNN, Graph Attention Network (GAT) that interprets it as a graph convolution method using the random walk normalized graph Laplacian. Using this framework, we theoretically prove that spectral sparsification preserves the features computed by the GAT model, thereby justifying our FastGAT algorithm. We experimentally evaluate FastGAT on several large real world graph datasets for node classification tasks, FastGAT can dramatically reduce (up to 10x) the computational time and memory requirements, allowing the usage of attention based GNNs on large graphs.
SkipGNN: Predicting Molecular Interactions with Skip-Graph Networks
Apr 30, 2020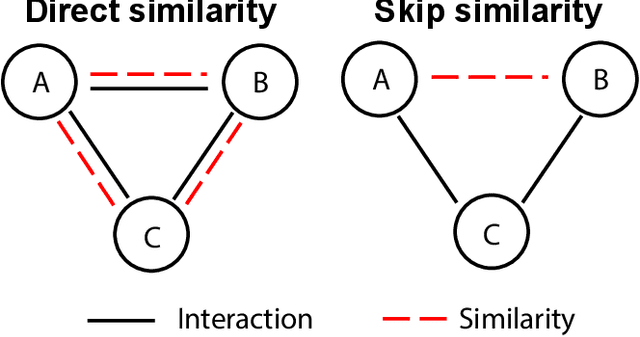
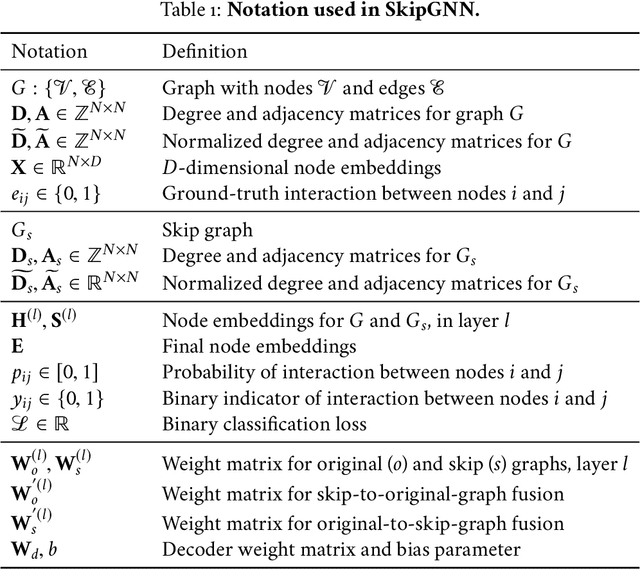
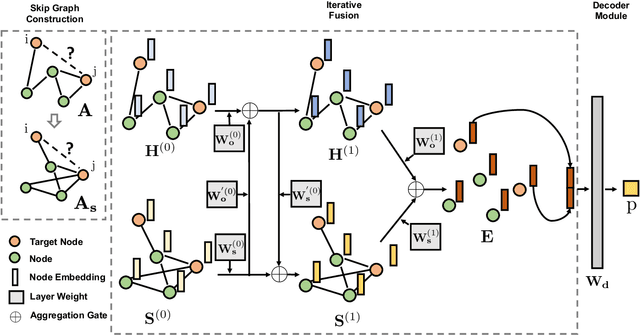
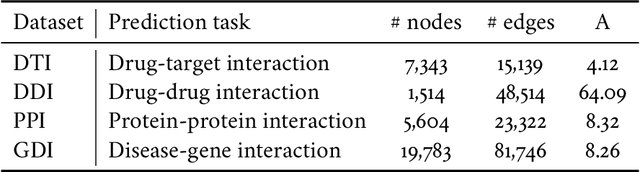
Molecular interaction networks are powerful resources for the discovery. They are increasingly used with machine learning methods to predict biologically meaningful interactions. While deep learning on graphs has dramatically advanced the prediction prowess, current graph neural network (GNN) methods are optimized for prediction on the basis of direct similarity between interacting nodes. In biological networks, however, similarity between nodes that do not directly interact has proved incredibly useful in the last decade across a variety of interaction networks. Here, we present SkipGNN, a graph neural network approach for the prediction of molecular interactions. SkipGNN predicts molecular interactions by not only aggregating information from direct interactions but also from second-order interactions, which we call skip similarity. In contrast to existing GNNs, SkipGNN receives neural messages from two-hop neighbors as well as immediate neighbors in the interaction network and non-linearly transforms the messages to obtain useful information for prediction. To inject skip similarity into a GNN, we construct a modified version of the original network, called the skip graph. We then develop an iterative fusion scheme that optimizes a GNN using both the skip graph and the original graph. Experiments on four interaction networks, including drug-drug, drug-target, protein-protein, and gene-disease interactions, show that SkipGNN achieves superior and robust performance, outperforming existing methods by up to 28.8\% of area under the precision recall curve (PR-AUC). Furthermore, we show that unlike popular GNNs, SkipGNN learns biologically meaningful embeddings and performs especially well on noisy, incomplete interaction networks.
MolTrans: Molecular Interaction Transformer for Drug Target Interaction Prediction
Apr 23, 2020
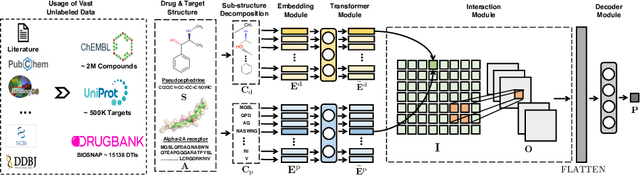
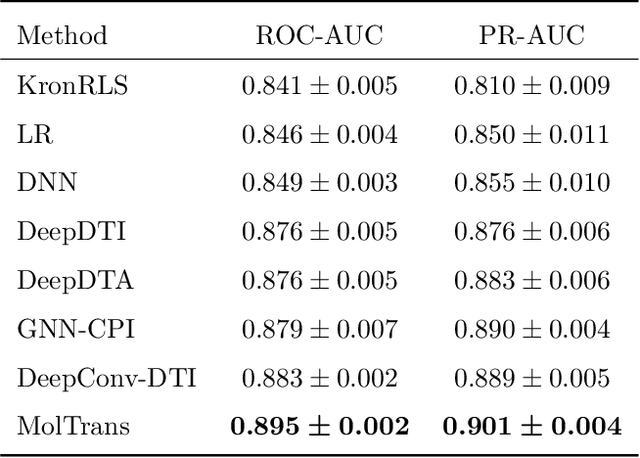
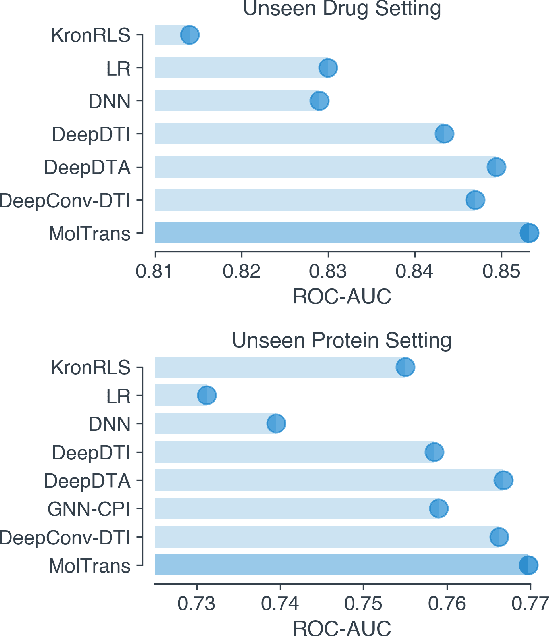
Drug target interaction (DTI) prediction is a foundational task for in silico drug discovery, which is costly and time-consuming due to the need of experimental search over large drug compound space. Recent years have witnessed promising progress for deep learning in DTI predictions. However, the following challenges are still open: (1) the sole data-driven molecular representation learning approaches ignore the sub-structural nature of DTI, thus produce results that are less accurate and difficult to explain; (2) existing methods focus on limited labeled data while ignoring the value of massive unlabelled molecular data. We propose a Molecular Interaction Transformer (MolTrans) to address these limitations via: (1) knowledge inspired sub-structural pattern mining algorithm and interaction modeling module for more accurate and interpretable DTI prediction; (2) an augmented transformer encoder to better extract and capture the semantic relations among substructures extracted from massive unlabeled biomedical data. We evaluate MolTrans on real world data and show it improved DTI prediction performance compared to state-of-the-art baselines.
DeepPurpose: a Deep Learning Based Drug Repurposing Toolkit
Apr 19, 2020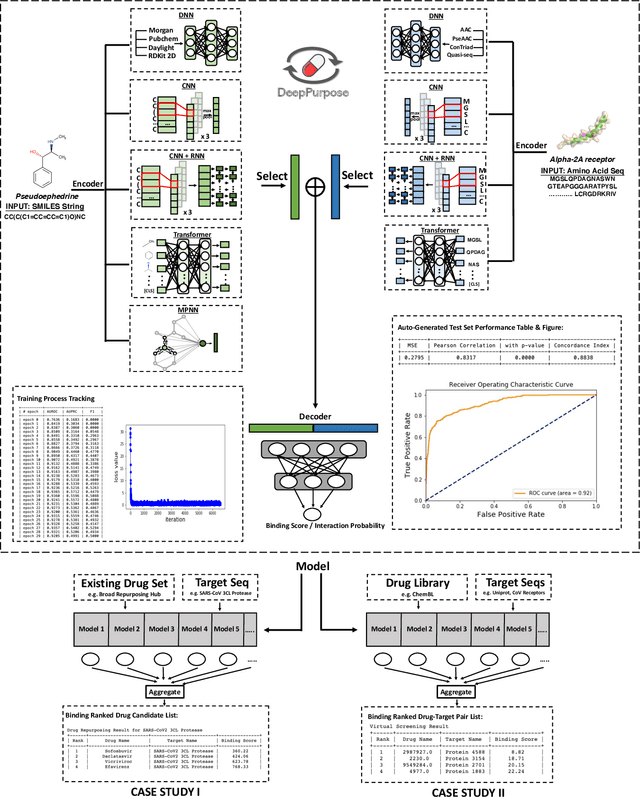
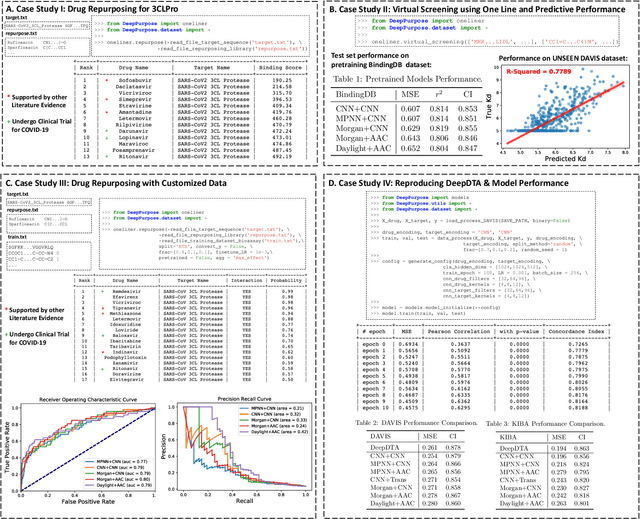
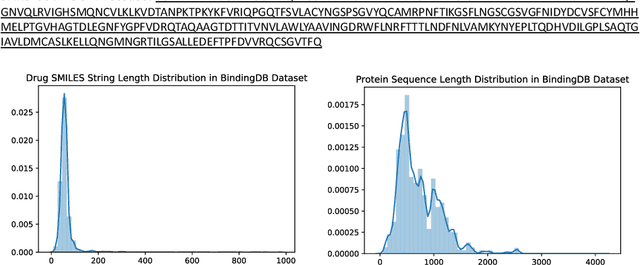
We present DeepPurpose, a deep learning toolkit for simple and efficient drug repurposing. With a few lines of code, DeepPurpose generates drug candidates based on aggregating five pretrained state-of-the-art models while offering flexibility for users to train their own models with 15 drug/target encodings and $50+$ novel architectures. We demonstrated DeepPurpose using case studies, including repurposing for COVID-19 where promising candidates under trials are ranked high in our results.
Predicting Treatment Initiation from Clinical Time Series Data via Graph-Augmented Time-Sensitive Model
Jul 01, 2019
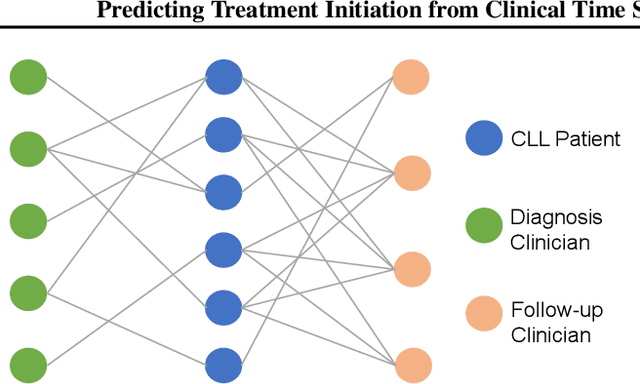
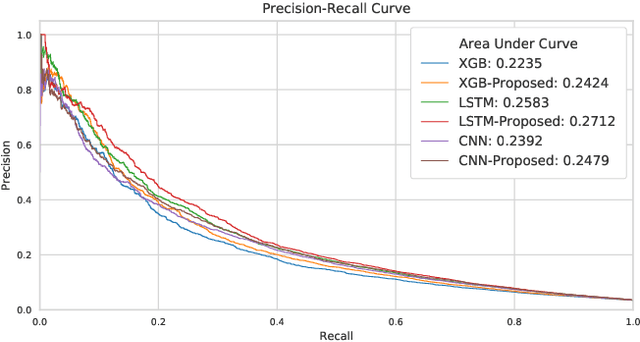
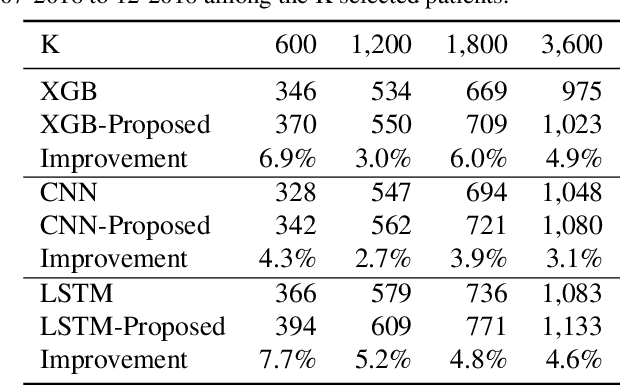
Many computational models were proposed to extract temporal patterns from clinical time series for each patient and among patient group for predictive healthcare. However, the common relations among patients (e.g., share the same doctor) were rarely considered. In this paper, we represent patients and clinicians relations by bipartite graphs addressing for example from whom a patient get a diagnosis. We then solve for the top eigenvectors of the graph Laplacian, and include the eigenvectors as latent representations of the similarity between patient-clinician pairs into a time-sensitive prediction model. We conducted experiments using real-world data to predict the initiation of first-line treatment for Chronic Lymphocytic Leukemia (CLL) patients. Results show that relational similarity can improve prediction over multiple baselines, for example a 5% incremental over long-short term memory baseline in terms of area under precision-recall curve.
Rare Disease Detection by Sequence Modeling with Generative Adversarial Networks
Jul 01, 2019
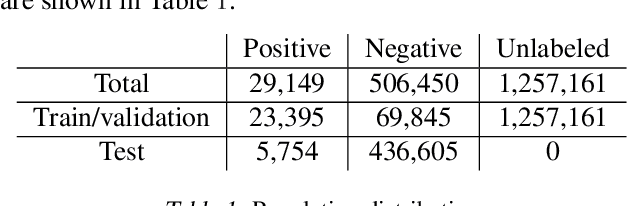


Rare diseases affecting 350 million individuals are commonly associated with delay in diagnosis or misdiagnosis. To improve those patients' outcome, rare disease detection is an important task for identifying patients with rare conditions based on longitudinal medical claims. In this paper, we present a deep learning method for detecting patients with exocrine pancreatic insufficiency (EPI) (a rare disease). The contribution includes 1) a large longitudinal study using 7 years medical claims from 1.8 million patients including 29,149 EPI patients, 2) a new deep learning model using generative adversarial networks (GANs) to boost rare disease class, and also leveraging recurrent neural networks to model patient sequence data, 3) an accurate prediction with 0.56 PR-AUC which outperformed benchmark models in terms of precision and recall.