 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAutomatic segmentation of spinal multiple sclerosis lesions: How to generalize across MRI contrasts?
Mar 11, 2020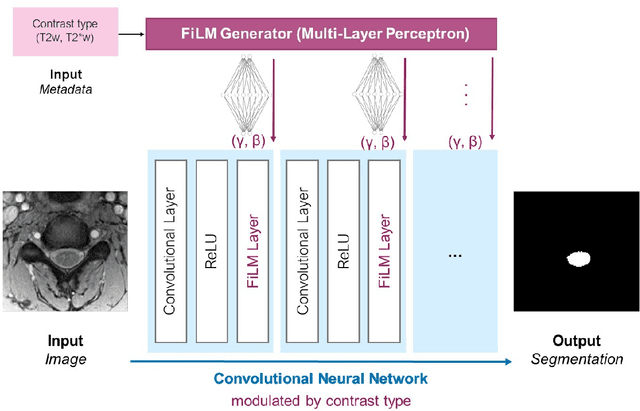
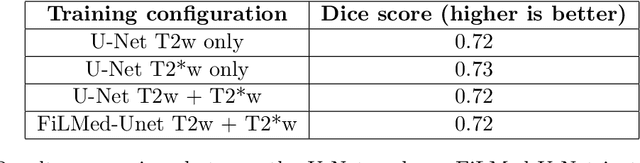
Despite recent improvements in medical image segmentation, the ability to generalize across imaging contrasts remains an open issue. To tackle this challenge, we implement Feature-wise Linear Modulation (FiLM) to leverage physics knowledge within the segmentation model and learn the characteristics of each contrast. Interestingly, a well-optimised U-Net reached the same performance as our FiLMed-Unet on a multi-contrast dataset (0.72 of Dice score), which suggests that there is a bottleneck in spinal MS lesion segmentation different from the generalization across varying contrasts. This bottleneck likely stems from inter-rater variability, which is estimated at 0.61 of Dice score in our dataset.
Quantifying the Value of Lateral Views in Deep Learning for Chest X-rays
Feb 07, 2020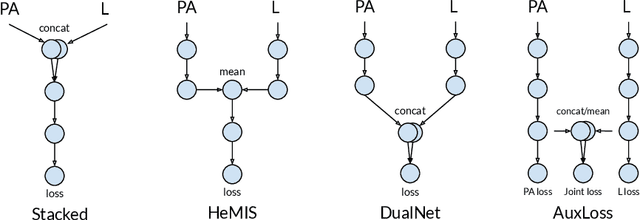
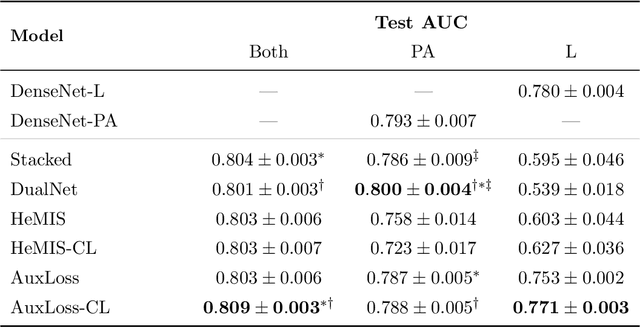
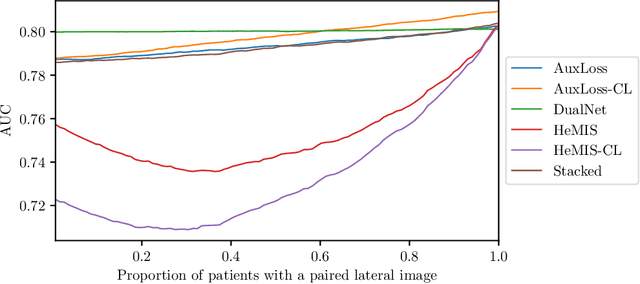
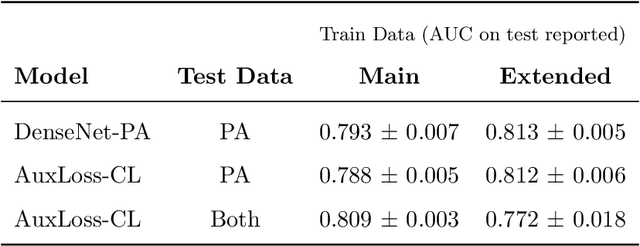
Most deep learning models in chest X-ray prediction utilize the posteroanterior (PA) view due to the lack of other views available. PadChest is a large-scale chest X-ray dataset that has almost 200 labels and multiple views available. In this work, we use PadChest to explore multiple approaches to merging the PA and lateral views for predicting the radiological labels associated with the X-ray image. We find that different methods of merging the model utilize the lateral view differently. We also find that including the lateral view increases performance for 32 labels in the dataset, while being neutral for the others. The increase in overall performance is comparable to the one obtained by using only the PA view with twice the amount of patients in the training set.
On the limits of cross-domain generalization in automated X-ray prediction
Feb 06, 2020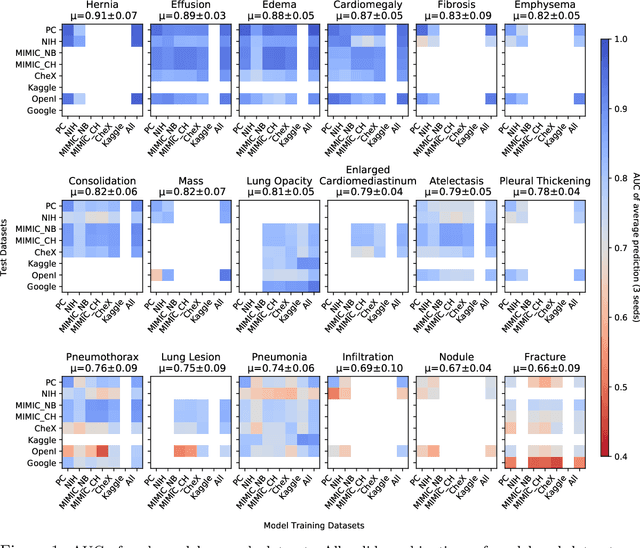
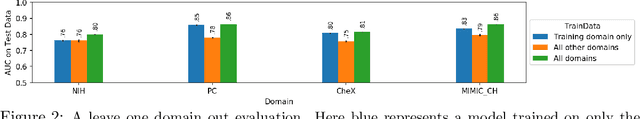
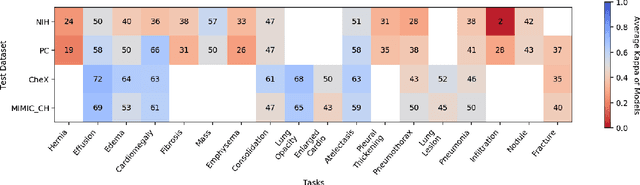
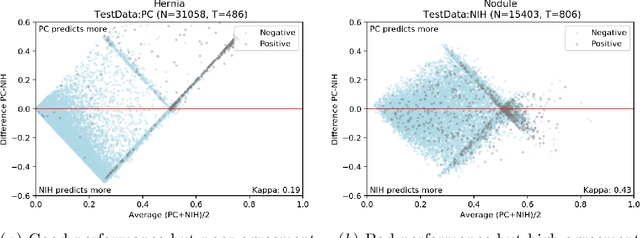
This large scale study focuses on quantifying what X-rays diagnostic prediction tasks generalize well across multiple different datasets. We present evidence that the issue of generalization is not due to a shift in the images but instead a shift in the labels. We study the cross-domain performance, agreement between models, and model representations. We find interesting discrepancies between performance and agreement where models which both achieve good performance disagree in their predictions as well as models which agree yet achieve poor performance. We also test for concept similarity by regularizing a network to group tasks across multiple datasets together and observe variation across the tasks.
Is graph-based feature selection of genes better than random?
Nov 19, 2019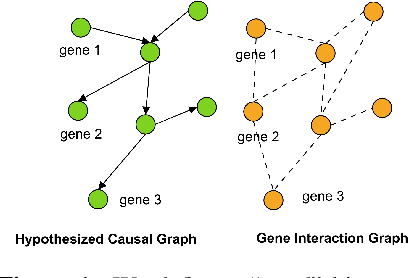
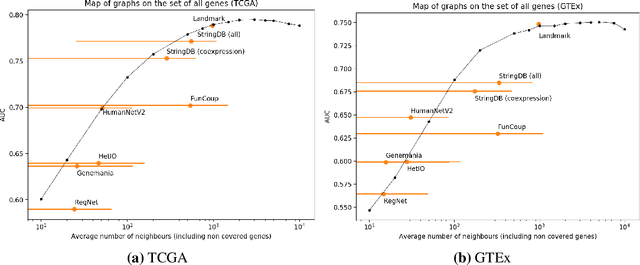
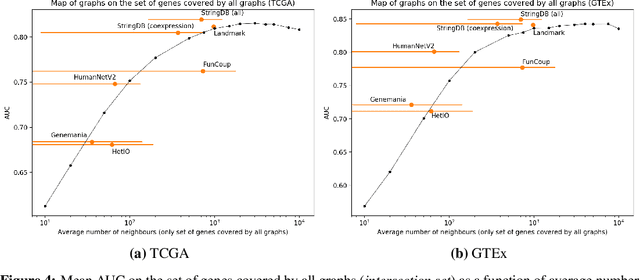
Gene interaction graphs aim to capture various relationships between genes and represent decades of biology research. When trying to make predictions from genomic data, those graphs could be used to overcome the curse of dimensionality by making machine learning models sparser and more consistent with biological common knowledge. In this work, we focus on assessing whether those graphs capture dependencies seen in gene expression data better than random. We formulate a condition that graphs should satisfy to provide a good prior knowledge and propose to test it using a `Single Gene Inference' (SGI) task. We compare random graphs with seven major gene interaction graphs published by different research groups, aiming to measure the true benefit of using biologically relevant graphs in this context. Our analysis finds that dependencies can be captured almost as well at random which suggests that, in terms of gene expression levels, the relevant information about the state of the cell is spread across many genes.
Deep Semantic Segmentation of Natural and Medical Images: A Review
Nov 02, 2019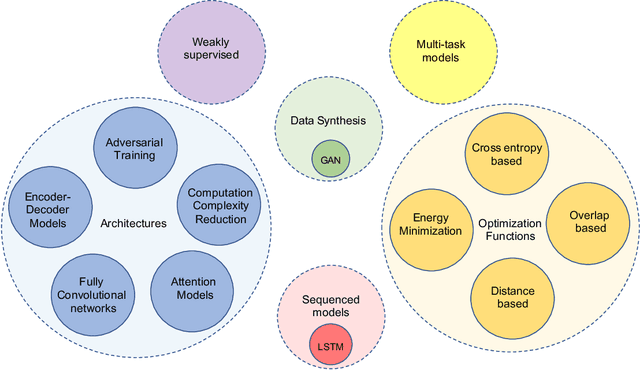
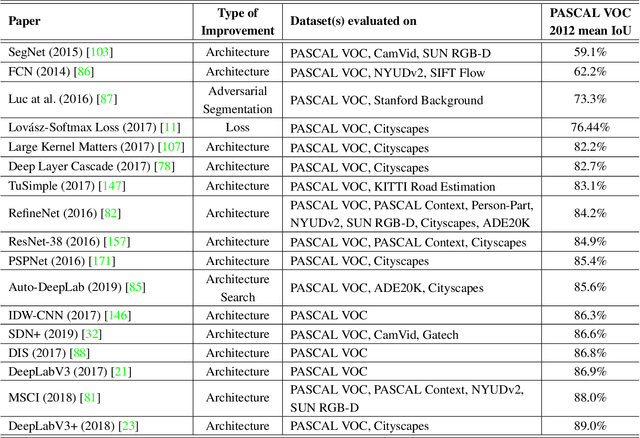
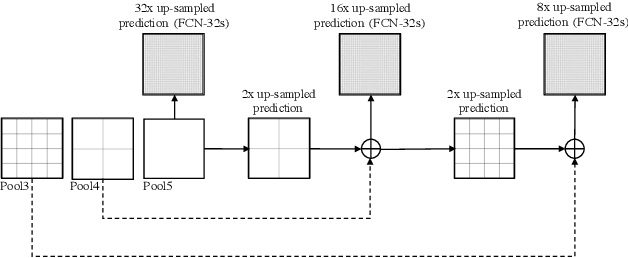
The (medical) image semantic segmentation task consists of classifying each pixel of an image (or just several ones) into an instance, where each instance (or category) corresponding to a class. This task is a part of the concept of scene understanding or better explaining the global context of an image. In the medical image analysis domain, image segmentation can be used for image-guided interventions, radiotherapy, or improved radiological diagnostics. In this review, we categorize the main deep learning-based medical and non-medical image segmentation solutions into six main groups of deep architectural improvements, data synthesis-based, loss function-based improvements, sequenced models, weakly supervised, and multi-task methods and further for each group we analyzed each variant of these groups and discuss limitations of the current approaches and future research directions for semantic image segmentation.
Icentia11K: An Unsupervised Representation Learning Dataset for Arrhythmia Subtype Discovery
Oct 21, 2019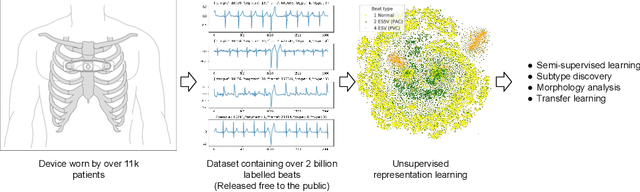
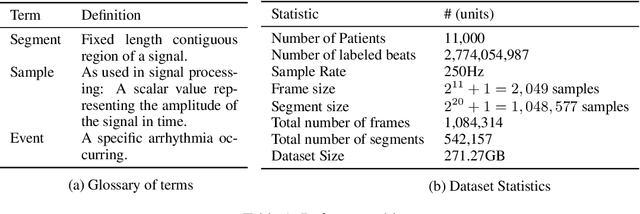
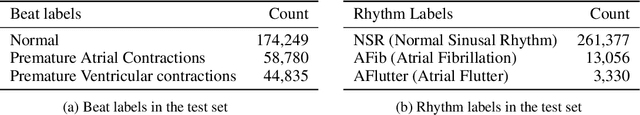
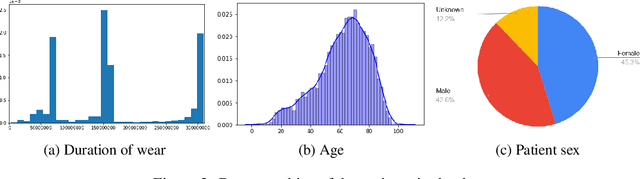
We release the largest public ECG dataset of continuous raw signals for representation learning containing 11 thousand patients and 2 billion labelled beats. Our goal is to enable semi-supervised ECG models to be made as well as to discover unknown subtypes of arrhythmia and anomalous ECG signal events. To this end, we propose an unsupervised representation learning task, evaluated in a semi-supervised fashion. We provide a set of baselines for different feature extractors that can be built upon. Additionally, we perform qualitative evaluations on results from PCA embeddings, where we identify some clustering of known subtypes indicating the potential for representation learning in arrhythmia sub-type discovery.
The TCGA Meta-Dataset Clinical Benchmark
Oct 18, 2019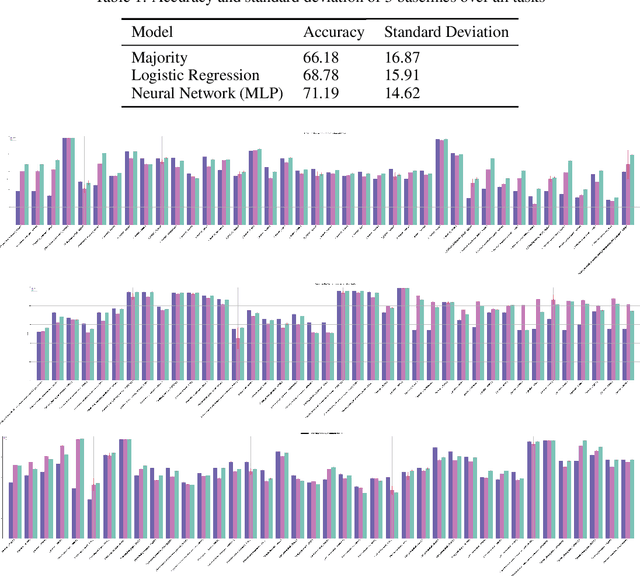
Machine learning is bringing a paradigm shift to healthcare by changing the process of disease diagnosis and prognosis in clinics and hospitals. This development equips doctors and medical staff with tools to evaluate their hypotheses and hence make more precise decisions. Although most current research in the literature seeks to develop techniques and methods for predicting one particular clinical outcome, this approach is far from the reality of clinical decision making in which you have to consider several factors simultaneously. In addition, it is difficult to follow the recent progress concretely as there is a lack of consistency in benchmark datasets and task definitions in the field of Genomics. To address the aforementioned issues, we provide a clinical Meta-Dataset derived from the publicly available data hub called The Cancer Genome Atlas Program (TCGA) that contains 174 tasks. We believe those tasks could be good proxy tasks to develop methods which can work on a few samples of gene expression data. Also, learning to predict multiple clinical variables using gene-expression data is an important task due to the variety of phenotypes in clinical problems and lack of samples for some of the rare variables. The defined tasks cover a wide range of clinical problems including predicting tumor tissue site, white cell count, histological type, family history of cancer, gender, and many others which we explain later in the paper. Each task represents an independent dataset. We use regression and neural network baselines for all the tasks using only 150 samples and compare their performance.
Underwhelming Generalization Improvements From Controlling Feature Attribution
Oct 01, 2019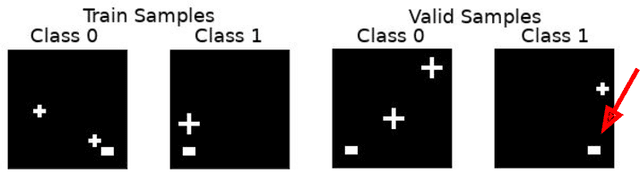
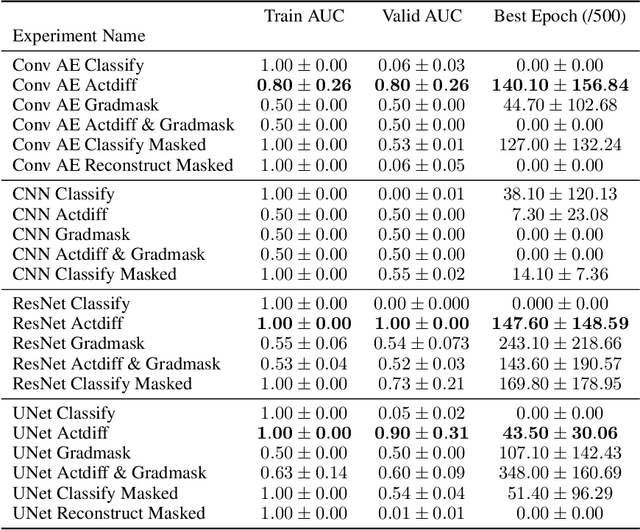
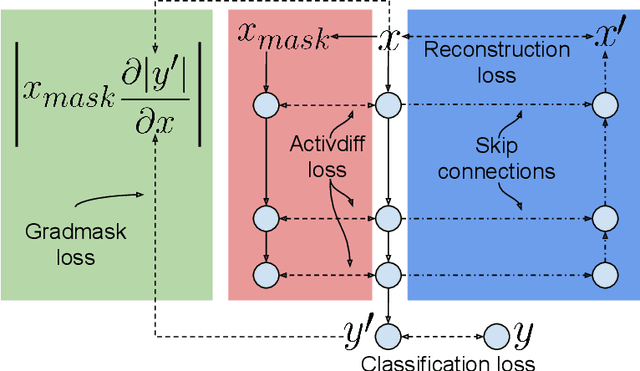
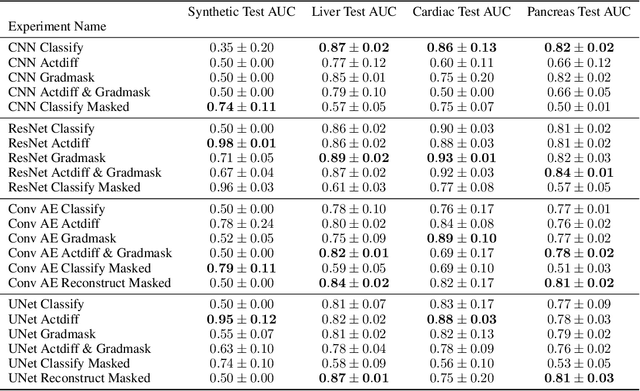
Overfitting is a common issue in machine learning, which can arise when the model learns to predict class membership using convenient but spuriously-correlated image features instead of the true image features that denote a class. These are typically visualized using saliency maps. In some object classification tasks such as for medical images, one may have some images with masks, indicating a region of interest, i.e., which part of the image contains the most relevant information for the classification. We describe a simple method for taking advantage of such auxiliary labels, by training networks to ignore the distracting features which may be extracted outside of the region of interest, on the training images for which such masks are available. This mask information is only used during training and has an impact on generalization accuracy in a dataset-dependent way. We observe an underwhelming relationship between controlling saliency maps and improving generalization performance.
Torchmeta: A Meta-Learning library for PyTorch
Sep 14, 2019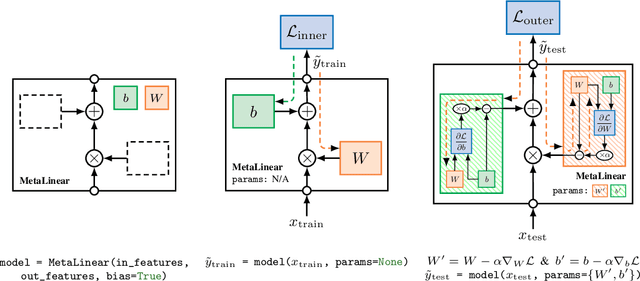
The constant introduction of standardized benchmarks in the literature has helped accelerating the recent advances in meta-learning research. They offer a way to get a fair comparison between different algorithms, and the wide range of datasets available allows full control over the complexity of this evaluation. However, for a large majority of code available online, the data pipeline is often specific to one dataset, and testing on another dataset requires significant rework. We introduce Torchmeta, a library built on top of PyTorch that enables seamless and consistent evaluation of meta-learning algorithms on multiple datasets, by providing data-loaders for most of the standard benchmarks in few-shot classification and regression, with a new meta-dataset abstraction. It also features some extensions for PyTorch to simplify the development of models compatible with meta-learning algorithms. The code is available here: https://github.com/tristandeleu/pytorch-meta
Analysis of Gene Interaction Graphs for Biasing Machine Learning Models
May 06, 2019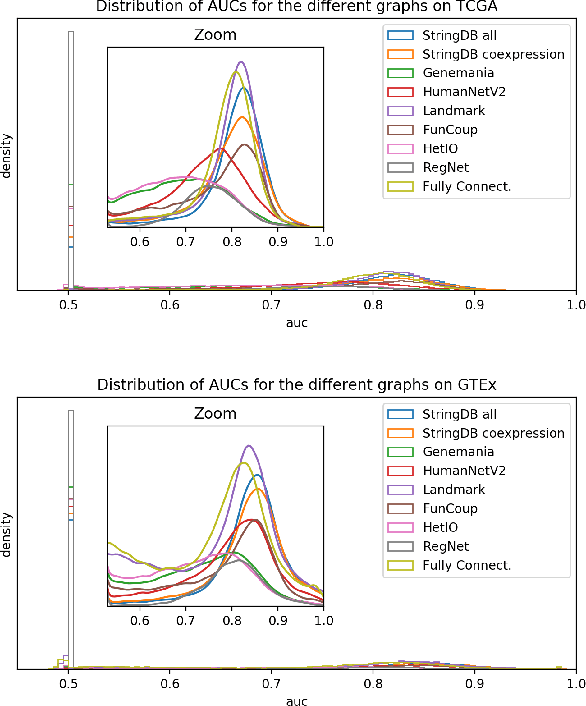
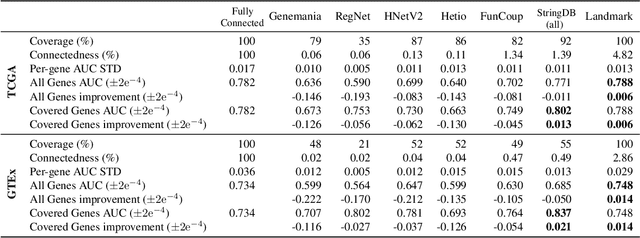
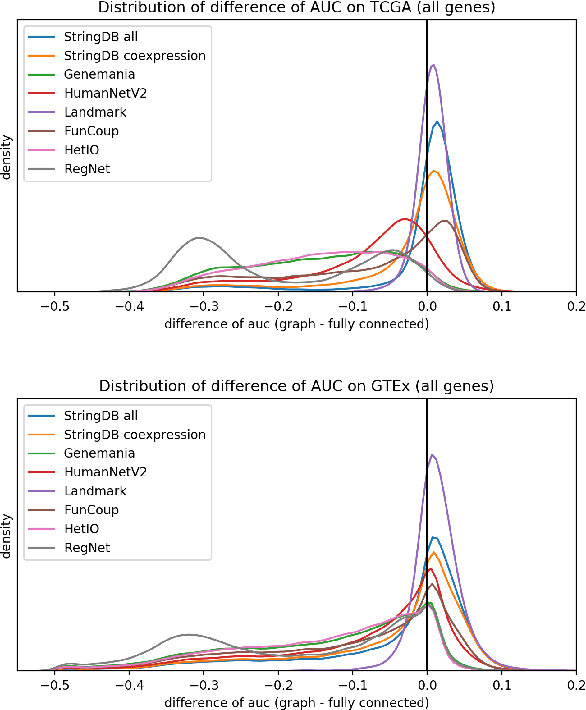
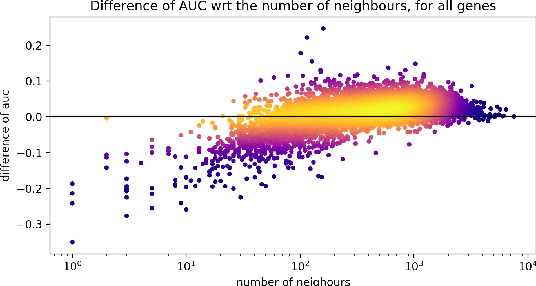
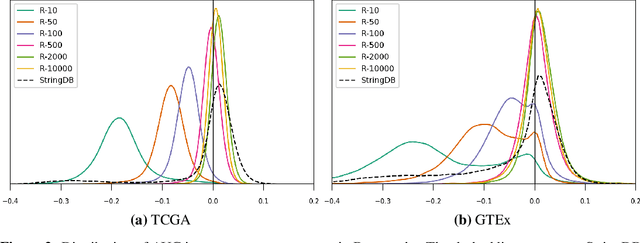
Gene interaction graphs aim to capture various relationships between genes and can be used to create more biologically-intuitive models for machine learning. There are many such graphs available which can differ in the number of genes and edges covered. In this work, we attempt to evaluate the biases provided by those graphs through utilizing them for 'Single Gene Inference' (SGI) which serves as, what we believe is, a proxy for more relevant prediction tasks. The SGI task assesses how well a gene's neighbors in a particular graph can 'explain' the gene itself in comparison to the baseline of using all the genes in the dataset. We evaluate seven major gene interaction graphs created by different research groups on two distinct datasets, TCGA and GTEx. We find that some graphs perform on par with the unbiased baseline for most genes with a significantly smaller feature set.