 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeFLOWR: Flow Matching for Structure-Aware De Novo, Interaction- and Fragment-Based Ligand Generation
Apr 14, 2025



We introduce FLOWR, a novel structure-based framework for the generation and optimization of three-dimensional ligands. FLOWR integrates continuous and categorical flow matching with equivariant optimal transport, enhanced by an efficient protein pocket conditioning. Alongside FLOWR, we present SPINDR, a thoroughly curated dataset comprising ligand-pocket co-crystal complexes specifically designed to address existing data quality issues. Empirical evaluations demonstrate that FLOWR surpasses current state-of-the-art diffusion- and flow-based methods in terms of PoseBusters-validity, pose accuracy, and interaction recovery, while offering a significant inference speedup, achieving up to 70-fold faster performance. In addition, we introduce FLOWR.multi, a highly accurate multi-purpose model allowing for the targeted sampling of novel ligands that adhere to predefined interaction profiles and chemical substructures for fragment-based design without the need of re-training or any re-sampling strategies
PepINVENT: Generative peptide design beyond the natural amino acids
Sep 21, 2024



Peptides play a crucial role in the drug design and discovery whether as a therapeutic modality or a delivery agent. Non-natural amino acids (NNAAs) have been used to enhance the peptide properties from binding affinity, plasma stability to permeability. Incorporating novel NNAAs facilitates the design of more effective peptides with improved properties. The generative models used in the field, have focused on navigating the peptide sequence space. The sequence space is formed by combinations of a predefined set of amino acids. However, there is still a need for a tool to explore the peptide landscape beyond this enumerated space to unlock and effectively incorporate de novo design of new amino acids. To thoroughly explore the theoretical chemical space of the peptides, we present PepINVENT, a novel generative AI-based tool as an extension to the small molecule molecular design platform, REINVENT. PepINVENT navigates the vast space of natural and non-natural amino acids to propose valid, novel, and diverse peptide designs. The generative model can serve as a central tool for peptide-related tasks, as it was not trained on peptides with specific properties or topologies. The prior was trained to understand the granularity of peptides and to design amino acids for filling the masked positions within a peptide. PepINVENT coupled with reinforcement learning enables the goal-oriented design of peptides using its chemistry-informed generative capabilities. This study demonstrates PepINVENT's ability to explore the peptide space with unique and novel designs, and its capacity for property optimization in the context of therapeutically relevant peptides. Our tool can be employed for multi-parameter learning objectives, peptidomimetics, lead optimization, and variety of other tasks within the peptide domain.
Masked Attention is All You Need for Graphs
Feb 16, 2024Graph neural networks (GNNs) and variations of the message passing algorithm are the predominant means for learning on graphs, largely due to their flexibility, speed, and satisfactory performance. The design of powerful and general purpose GNNs, however, requires significant research efforts and often relies on handcrafted, carefully-chosen message passing operators. Motivated by this, we propose a remarkably simple alternative for learning on graphs that relies exclusively on attention. Graphs are represented as node or edge sets and their connectivity is enforced by masking the attention weight matrix, effectively creating custom attention patterns for each graph. Despite its simplicity, masked attention for graphs (MAG) has state-of-the-art performance on long-range tasks and outperforms strong message passing baselines and much more involved attention-based methods on over 55 node and graph-level tasks. We also show significantly better transfer learning capabilities compared to GNNs and comparable or better time and memory scaling. MAG has sub-linear memory scaling in the number of nodes or edges, enabling learning on dense graphs and future-proofing the approach.
Graph Neural Networks with Adaptive Readouts
Nov 09, 2022An effective aggregation of node features into a graph-level representation via readout functions is an essential step in numerous learning tasks involving graph neural networks. Typically, readouts are simple and non-adaptive functions designed such that the resulting hypothesis space is permutation invariant. Prior work on deep sets indicates that such readouts might require complex node embeddings that can be difficult to learn via standard neighborhood aggregation schemes. Motivated by this, we investigate the potential of adaptive readouts given by neural networks that do not necessarily give rise to permutation invariant hypothesis spaces. We argue that in some problems such as binding affinity prediction where molecules are typically presented in a canonical form it might be possible to relax the constraints on permutation invariance of the hypothesis space and learn a more effective model of the affinity by employing an adaptive readout function. Our empirical results demonstrate the effectiveness of neural readouts on more than 40 datasets spanning different domains and graph characteristics. Moreover, we observe a consistent improvement over standard readouts (i.e., sum, max, and mean) relative to the number of neighborhood aggregation iterations and different convolutional operators.
Representations and Strategies for Transferable Machine Learning Models in Chemical Discovery
Jun 20, 2021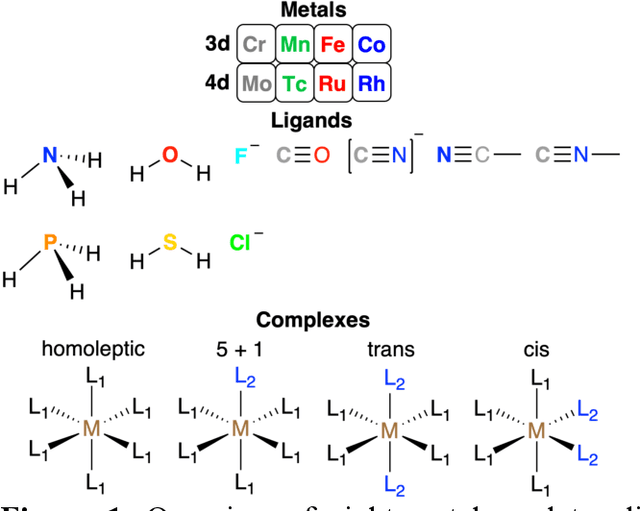
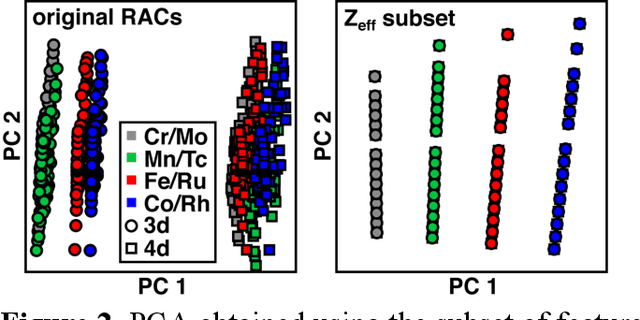
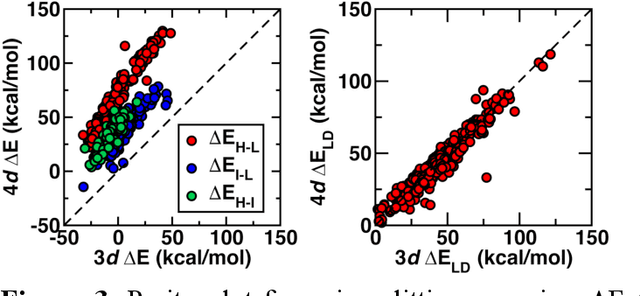
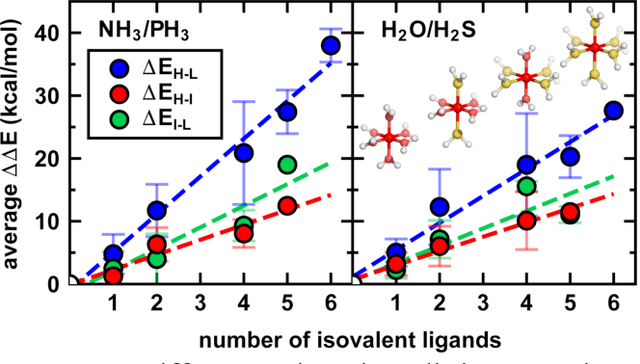
Strategies for machine-learning(ML)-accelerated discovery that are general across materials composition spaces are essential, but demonstrations of ML have been primarily limited to narrow composition variations. By addressing the scarcity of data in promising regions of chemical space for challenging targets like open-shell transition-metal complexes, general representations and transferable ML models that leverage known relationships in existing data will accelerate discovery. Over a large set (ca. 1000) of isovalent transition-metal complexes, we quantify evident relationships for different properties (i.e., spin-splitting and ligand dissociation) between rows of the periodic table (i.e., 3d/4d metals and 2p/3p ligands). We demonstrate an extension to graph-based revised autocorrelation (RAC) representation (i.e., eRAC) that incorporates the effective nuclear charge alongside the nuclear charge heuristic that otherwise overestimates dissimilarity of isovalent complexes. To address the common challenge of discovery in a new space where data is limited, we introduce a transfer learning approach in which we seed models trained on a large amount of data from one row of the periodic table with a small number of data points from the additional row. We demonstrate the synergistic value of the eRACs alongside this transfer learning strategy to consistently improve model performance. Analysis of these models highlights how the approach succeeds by reordering the distances between complexes to be more consistent with the periodic table, a property we expect to be broadly useful for other materials domains.