 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Transferable Recommender Approach for Selecting the Best Density Functional Approximations in Chemical Discovery
Jul 21, 2022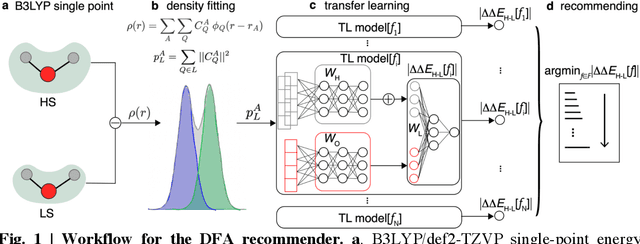
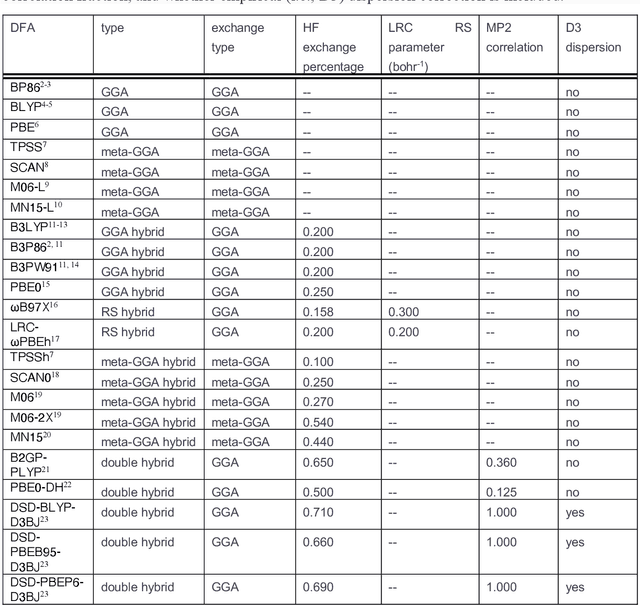
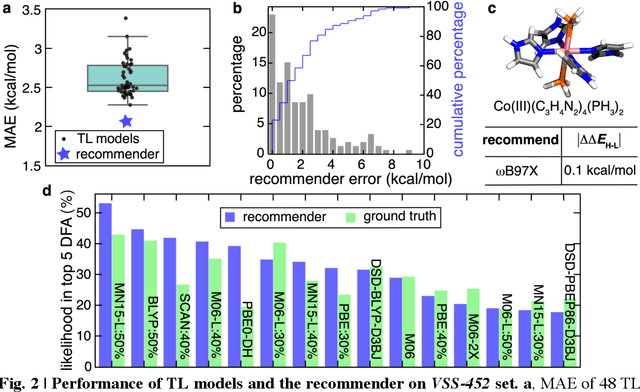
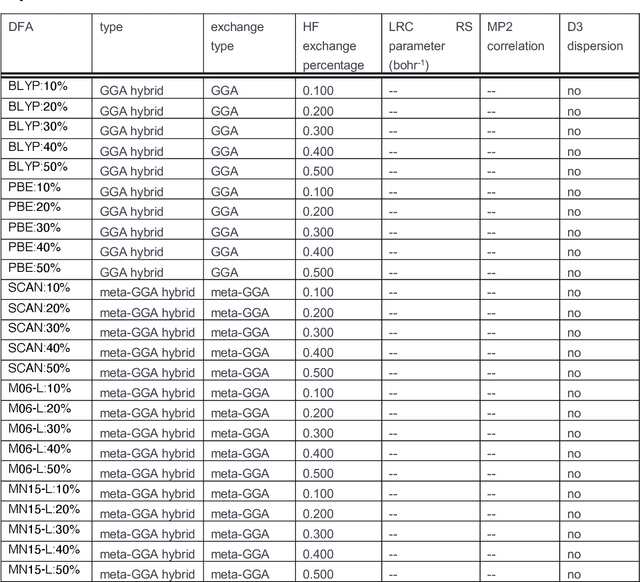
Approximate density functional theory (DFT) has become indispensable owing to its cost-accuracy trade-off in comparison to more computationally demanding but accurate correlated wavefunction theory. To date, however, no single density functional approximation (DFA) with universal accuracy has been identified, leading to uncertainty in the quality of data generated from DFT. With electron density fitting and transfer learning, we build a DFA recommender that selects the DFA with the lowest expected error with respect to gold standard but cost-prohibitive coupled cluster theory in a system-specific manner. We demonstrate this recommender approach on vertical spin-splitting energy evaluation for challenging transition metal complexes. Our recommender predicts top-performing DFAs and yields excellent accuracy (ca. 2 kcal/mol) for chemical discovery, outperforming both individual transfer learning models and the single best functional in a set of 48 DFAs. We demonstrate the transferability of the DFA recommender to experimentally synthesized compounds with distinct chemistry.
Representations and Strategies for Transferable Machine Learning Models in Chemical Discovery
Jun 20, 2021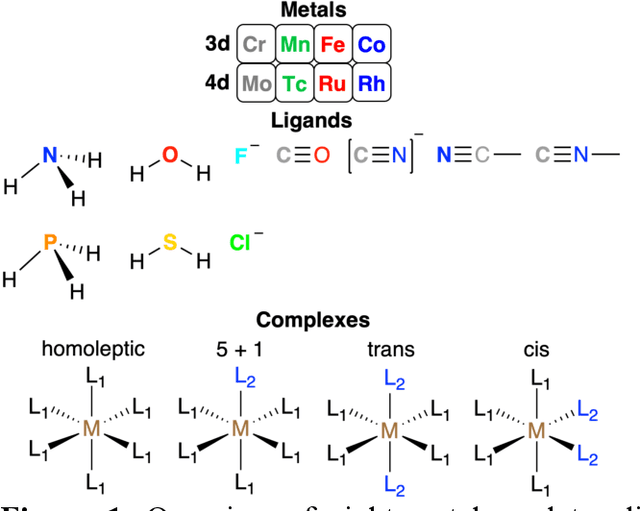
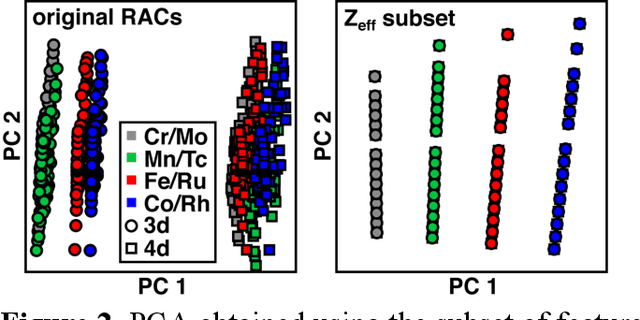
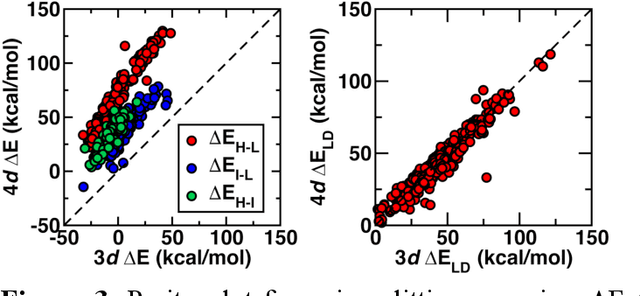
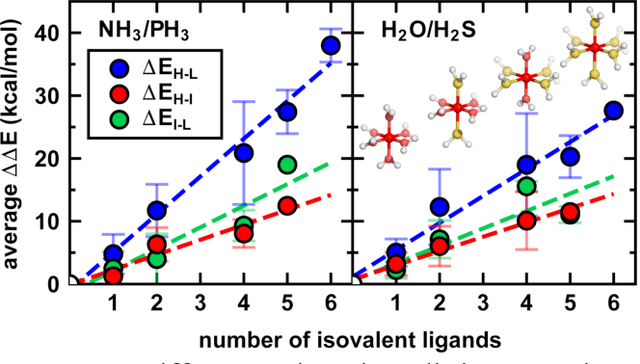
Strategies for machine-learning(ML)-accelerated discovery that are general across materials composition spaces are essential, but demonstrations of ML have been primarily limited to narrow composition variations. By addressing the scarcity of data in promising regions of chemical space for challenging targets like open-shell transition-metal complexes, general representations and transferable ML models that leverage known relationships in existing data will accelerate discovery. Over a large set (ca. 1000) of isovalent transition-metal complexes, we quantify evident relationships for different properties (i.e., spin-splitting and ligand dissociation) between rows of the periodic table (i.e., 3d/4d metals and 2p/3p ligands). We demonstrate an extension to graph-based revised autocorrelation (RAC) representation (i.e., eRAC) that incorporates the effective nuclear charge alongside the nuclear charge heuristic that otherwise overestimates dissimilarity of isovalent complexes. To address the common challenge of discovery in a new space where data is limited, we introduce a transfer learning approach in which we seed models trained on a large amount of data from one row of the periodic table with a small number of data points from the additional row. We demonstrate the synergistic value of the eRACs alongside this transfer learning strategy to consistently improve model performance. Analysis of these models highlights how the approach succeeds by reordering the distances between complexes to be more consistent with the periodic table, a property we expect to be broadly useful for other materials domains.