 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeLearned representation-guided diffusion models for large-image generation
Dec 12, 2023



To synthesize high-fidelity samples, diffusion models typically require auxiliary data to guide the generation process. However, it is impractical to procure the painstaking patch-level annotation effort required in specialized domains like histopathology and satellite imagery; it is often performed by domain experts and involves hundreds of millions of patches. Modern-day self-supervised learning (SSL) representations encode rich semantic and visual information. In this paper, we posit that such representations are expressive enough to act as proxies to fine-grained human labels. We introduce a novel approach that trains diffusion models conditioned on embeddings from SSL. Our diffusion models successfully project these features back to high-quality histopathology and remote sensing images. In addition, we construct larger images by assembling spatially consistent patches inferred from SSL embeddings, preserving long-range dependencies. Augmenting real data by generating variations of real images improves downstream classifier accuracy for patch-level and larger, image-scale classification tasks. Our models are effective even on datasets not encountered during training, demonstrating their robustness and generalizability. Generating images from learned embeddings is agnostic to the source of the embeddings. The SSL embeddings used to generate a large image can either be extracted from a reference image, or sampled from an auxiliary model conditioned on any related modality (e.g. class labels, text, genomic data). As proof of concept, we introduce the text-to-large image synthesis paradigm where we successfully synthesize large pathology and satellite images out of text descriptions.
Attention De-sparsification Matters: Inducing Diversity in Digital Pathology Representation Learning
Sep 12, 2023



We propose DiRL, a Diversity-inducing Representation Learning technique for histopathology imaging. Self-supervised learning techniques, such as contrastive and non-contrastive approaches, have been shown to learn rich and effective representations of digitized tissue samples with limited pathologist supervision. Our analysis of vanilla SSL-pretrained models' attention distribution reveals an insightful observation: sparsity in attention, i.e, models tends to localize most of their attention to some prominent patterns in the image. Although attention sparsity can be beneficial in natural images due to these prominent patterns being the object of interest itself, this can be sub-optimal in digital pathology; this is because, unlike natural images, digital pathology scans are not object-centric, but rather a complex phenotype of various spatially intermixed biological components. Inadequate diversification of attention in these complex images could result in crucial information loss. To address this, we leverage cell segmentation to densely extract multiple histopathology-specific representations, and then propose a prior-guided dense pretext task for SSL, designed to match the multiple corresponding representations between the views. Through this, the model learns to attend to various components more closely and evenly, thus inducing adequate diversification in attention for capturing context rich representations. Through quantitative and qualitative analysis on multiple tasks across cancer types, we demonstrate the efficacy of our method and observe that the attention is more globally distributed.
PathLDM: Text conditioned Latent Diffusion Model for Histopathology
Sep 01, 2023



To achieve high-quality results, diffusion models must be trained on large datasets. This can be notably prohibitive for models in specialized domains, such as computational pathology. Conditioning on labeled data is known to help in data-efficient model training. Therefore, histopathology reports, which are rich in valuable clinical information, are an ideal choice as guidance for a histopathology generative model. In this paper, we introduce PathLDM, the first text-conditioned Latent Diffusion Model tailored for generating high-quality histopathology images. Leveraging the rich contextual information provided by pathology text reports, our approach fuses image and textual data to enhance the generation process. By utilizing GPT's capabilities to distill and summarize complex text reports, we establish an effective conditioning mechanism. Through strategic conditioning and necessary architectural enhancements, we achieved a SoTA FID score of 7.64 for text-to-image generation on the TCGA-BRCA dataset, significantly outperforming the closest text-conditioned competitor with FID 30.1.
SAM-Path: A Segment Anything Model for Semantic Segmentation in Digital Pathology
Jul 12, 2023



Semantic segmentations of pathological entities have crucial clinical value in computational pathology workflows. Foundation models, such as the Segment Anything Model (SAM), have been recently proposed for universal use in segmentation tasks. SAM shows remarkable promise in instance segmentation on natural images. However, the applicability of SAM to computational pathology tasks is limited due to the following factors: (1) lack of comprehensive pathology datasets used in SAM training and (2) the design of SAM is not inherently optimized for semantic segmentation tasks. In this work, we adapt SAM for semantic segmentation by introducing trainable class prompts, followed by further enhancements through the incorporation of a pathology encoder, specifically a pathology foundation model. Our framework, SAM-Path enhances SAM's ability to conduct semantic segmentation in digital pathology without human input prompts. Through experiments on two public pathology datasets, the BCSS and the CRAG datasets, we demonstrate that the fine-tuning with trainable class prompts outperforms vanilla SAM with manual prompts and post-processing by 27.52% in Dice score and 71.63% in IOU. On these two datasets, the proposed additional pathology foundation model further achieves a relative improvement of 5.07% to 5.12% in Dice score and 4.50% to 8.48% in IOU.
Halcyon -- A Pathology Imaging and Feature analysis and Management System
Apr 07, 2023



Halcyon is a new pathology imaging analysis and feature management system based on W3C linked-data open standards and is designed to scale to support the needs for the voluminous production of features from deep-learning feature pipelines. Halcyon can support multiple users with a web-based UX with access to all user data over a standards-based web API allowing for integration with other processes and software systems. Identity management and data security is also provided.
Topology-Guided Multi-Class Cell Context Generation for Digital Pathology
Apr 05, 2023



In digital pathology, the spatial context of cells is important for cell classification, cancer diagnosis and prognosis. To model such complex cell context, however, is challenging. Cells form different mixtures, lineages, clusters and holes. To model such structural patterns in a learnable fashion, we introduce several mathematical tools from spatial statistics and topological data analysis. We incorporate such structural descriptors into a deep generative model as both conditional inputs and a differentiable loss. This way, we are able to generate high quality multi-class cell layouts for the first time. We show that the topology-rich cell layouts can be used for data augmentation and improve the performance of downstream tasks such as cell classification.
Prompt-MIL: Boosting Multi-Instance Learning Schemes via Task-specific Prompt Tuning
Mar 21, 2023Whole slide image (WSI) classification is a critical task in computational pathology, requiring the processing of gigapixel-sized images, which is challenging for current deep-learning methods. Current state of the art methods are based on multi-instance learning schemes (MIL), which usually rely on pretrained features to represent the instances. Due to the lack of task-specific annotated data, these features are either obtained from well-established backbones on natural images, or, more recently from self-supervised models pretrained on histopathology. However, both approaches yield task-agnostic features, resulting in performance loss compared to the appropriate task-related supervision, if available. In this paper, we show that when task-specific annotations are limited, we can inject such supervision into downstream task training, to reduce the gap between fully task-tuned and task agnostic features. We propose Prompt-MIL, an MIL framework that integrates prompts into WSI classification. Prompt-MIL adopts a prompt tuning mechanism, where only a small fraction of parameters calibrates the pretrained features to encode task-specific information, rather than the conventional full fine-tuning approaches. Extensive experiments on three WSI datasets, TCGA-BRCA, TCGA-CRC, and BRIGHT, demonstrate the superiority of Prompt-MIL over conventional MIL methods, achieving a relative improvement of 1.49%-4.03% in accuracy and 0.25%-8.97% in AUROC while using fewer than 0.3% additional parameters. Compared to conventional full fine-tuning approaches, we fine-tune less than 1.3% of the parameters, yet achieve a relative improvement of 1.29%-13.61% in accuracy and 3.22%-27.18% in AUROC and reduce GPU memory consumption by 38%-45% while training 21%-27% faster.
Precise Location Matching Improves Dense Contrastive Learning in Digital Pathology
Dec 23, 2022Dense prediction tasks such as segmentation and detection of pathological entities hold crucial clinical value in the digital pathology workflow. However, obtaining dense annotations on large cohorts is usually tedious and expensive. Contrastive learning (CL) is thus often employed to leverage large volumes of unlabeled data to pre-train the backbone network. To boost CL for dense prediction, some studies have proposed variations of dense matching objectives in pre-training. However, our analysis shows that employing existing dense matching strategies on histopathology images enforces invariance among incorrect pairs of dense features and, thus, is imprecise. To address this, we propose a precise location-based matching mechanism that utilizes the overlapping information between geometric transformations to precisely match regions in two augmentations. Extensive experiments on two pretraining datasets (TCGA-BRCA, NCT-CRC-HE) and three downstream datasets (GlaS, CRAG, BCSS) highlight the superiority of our method in semantic and instance segmentation tasks. Our method outperforms previous dense matching methods by up to 7.2 % in average precision for detection and 5.6 % in average precision for instance segmentation tasks. Additionally, by using our matching mechanism in the three popular contrastive learning frameworks, MoCo-v2, VICRegL and ConCL, the average precision in detection is improved by 0.7 % to 5.2 % and the average precision in segmentation is improved by 0.7 % to 4.0 %, demonstrating its generalizability.
Learning Topological Interactions for Multi-Class Medical Image Segmentation
Jul 20, 2022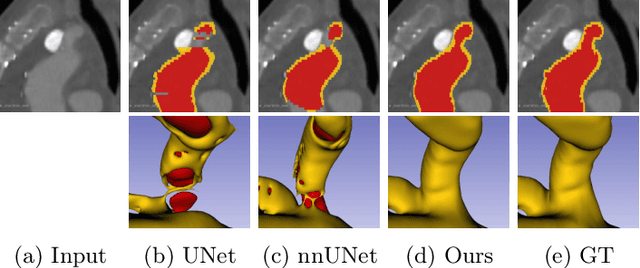
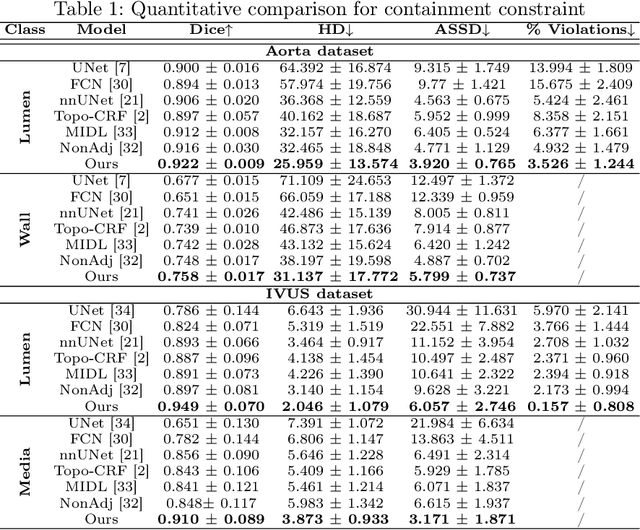
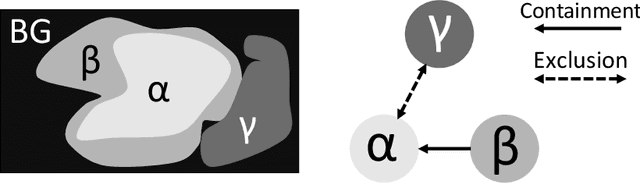
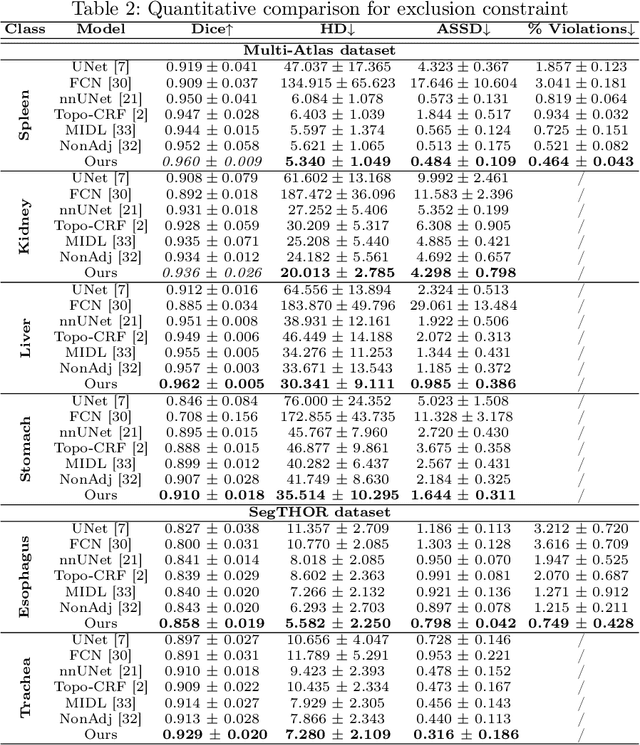
Deep learning methods have achieved impressive performance for multi-class medical image segmentation. However, they are limited in their ability to encode topological interactions among different classes (e.g., containment and exclusion). These constraints naturally arise in biomedical images and can be crucial in improving segmentation quality. In this paper, we introduce a novel topological interaction module to encode the topological interactions into a deep neural network. The implementation is completely convolution-based and thus can be very efficient. This empowers us to incorporate the constraints into end-to-end training and enrich the feature representation of neural networks. The efficacy of the proposed method is validated on different types of interactions. We also demonstrate the generalizability of the method on both proprietary and public challenge datasets, in both 2D and 3D settings, as well as across different modalities such as CT and Ultrasound. Code is available at: https://github.com/TopoXLab/TopoInteraction
Gigapixel Whole-Slide Images Classification using Locally Supervised Learning
Jul 17, 2022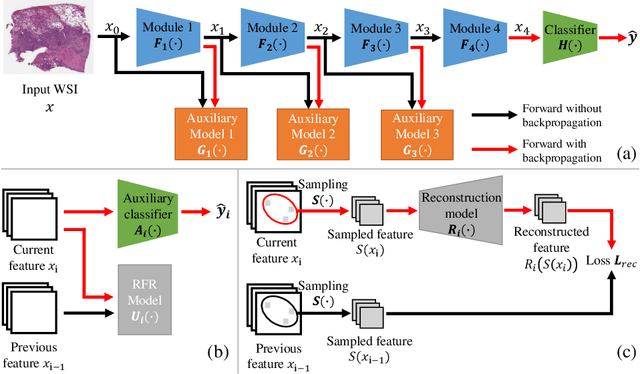
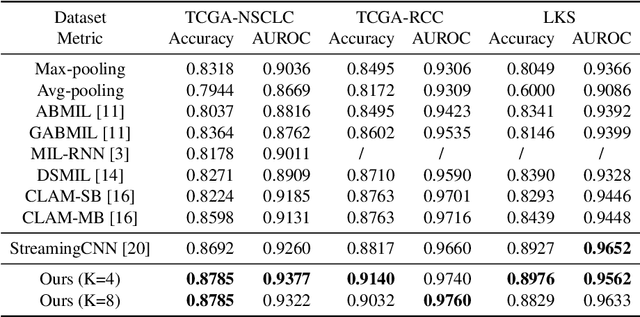


Histopathology whole slide images (WSIs) play a very important role in clinical studies and serve as the gold standard for many cancer diagnoses. However, generating automatic tools for processing WSIs is challenging due to their enormous sizes. Currently, to deal with this issue, conventional methods rely on a multiple instance learning (MIL) strategy to process a WSI at patch level. Although effective, such methods are computationally expensive, because tiling a WSI into patches takes time and does not explore the spatial relations between these tiles. To tackle these limitations, we propose a locally supervised learning framework which processes the entire slide by exploring the entire local and global information that it contains. This framework divides a pre-trained network into several modules and optimizes each module locally using an auxiliary model. We also introduce a random feature reconstruction unit (RFR) to preserve distinguishing features during training and improve the performance of our method by 1% to 3%. Extensive experiments on three publicly available WSI datasets: TCGA-NSCLC, TCGA-RCC and LKS, highlight the superiority of our method on different classification tasks. Our method outperforms the state-of-the-art MIL methods by 2% to 5% in accuracy, while being 7 to 10 times faster. Additionally, when dividing it into eight modules, our method requires as little as 20% of the total gpu memory required by end-to-end training. Our code is available at https://github.com/cvlab-stonybrook/local_learning_wsi.