 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMESS: Modern Electronic Structure Simulations
Jun 05, 2024Electronic structure simulation (ESS) has been used for decades to provide quantitative scientific insights on an atomistic scale, enabling advances in chemistry, biology, and materials science, among other disciplines. Following standard practice in scientific computing, the software packages driving these studies have been implemented in compiled languages such as FORTRAN and C. However, the recent introduction of machine learning (ML) into these domains has meant that ML models must be coded in these languages, or that complex software bridges have to be built between ML models in Python and these large compiled software systems. This is in contrast with recent progress in modern ML frameworks which aim to optimise both ease of use and high performance by harnessing hardware acceleration of tensor programs defined in Python. We introduce MESS: a modern electronic structure simulation package implemented in JAX; porting the ESS code to the ML world. We outline the costs and benefits of following the software development practices used in ML for this important scientific workload. MESS shows significant speedups n widely available hardware accelerators and simultaneously opens a clear pathway towards combining ESS with ML. MESS is available at https://github.com/graphcore-research/mess.
Reducing the Cost of Quantum Chemical Data By Backpropagating Through Density Functional Theory
Feb 06, 2024Density Functional Theory (DFT) accurately predicts the quantum chemical properties of molecules, but scales as $O(N_{\text{electrons}}^3)$. Sch\"utt et al. (2019) successfully approximate DFT 1000x faster with Neural Networks (NN). Arguably, the biggest problem one faces when scaling to larger molecules is the cost of DFT labels. For example, it took years to create the PCQ dataset (Nakata & Shimazaki, 2017) on which subsequent NNs are trained within a week. DFT labels molecules by minimizing energy $E(\cdot )$ as a "loss function." We bypass dataset creation by directly training NNs with $E(\cdot )$ as a loss function. For comparison, Sch\"utt et al. (2019) spent 626 hours creating a dataset on which they trained their NN for 160h, for a total of 786h; our method achieves comparable performance within 31h.
Generating QM1B with PySCF$_{\text{IPU}}$
Nov 02, 2023The emergence of foundation models in Computer Vision and Natural Language Processing have resulted in immense progress on downstream tasks. This progress was enabled by datasets with billions of training examples. Similar benefits are yet to be unlocked for quantum chemistry, where the potential of deep learning is constrained by comparatively small datasets with 100k to 20M training examples. These datasets are limited in size because the labels are computed using the accurate (but computationally demanding) predictions of Density Functional Theory (DFT). Notably, prior DFT datasets were created using CPU supercomputers without leveraging hardware acceleration. In this paper, we take a first step towards utilising hardware accelerators by introducing the data generator PySCF$_{\text{IPU}}$ using Intelligence Processing Units (IPUs). This allowed us to create the dataset QM1B with one billion training examples containing 9-11 heavy atoms. We demonstrate that a simple baseline neural network (SchNet 9M) improves its performance by simply increasing the amount of training data without additional inductive biases. To encourage future researchers to use QM1B responsibly, we highlight several limitations of QM1B and emphasise the low-resolution of our DFT options, which also serves as motivation for even larger, more accurate datasets. Code and dataset are available on Github: http://github.com/graphcore-research/pyscf-ipu
GPS++: Reviving the Art of Message Passing for Molecular Property Prediction
Feb 06, 2023



We present GPS++, a hybrid Message Passing Neural Network / Graph Transformer model for molecular property prediction. Our model integrates a well-tuned local message passing component and biased global attention with other key ideas from prior literature to achieve state-of-the-art results on large-scale molecular dataset PCQM4Mv2. Through a thorough ablation study we highlight the impact of individual components and, contrary to expectations set by recent trends, find that nearly all of the model's performance can be maintained without any use of global self-attention. We also show that our approach is significantly more accurate than prior art when 3D positional information is not available.
GPS++: An Optimised Hybrid MPNN/Transformer for Molecular Property Prediction
Dec 06, 2022



This technical report presents GPS++, the first-place solution to the Open Graph Benchmark Large-Scale Challenge (OGB-LSC 2022) for the PCQM4Mv2 molecular property prediction task. Our approach implements several key principles from the prior literature. At its core our GPS++ method is a hybrid MPNN/Transformer model that incorporates 3D atom positions and an auxiliary denoising task. The effectiveness of GPS++ is demonstrated by achieving 0.0719 mean absolute error on the independent test-challenge PCQM4Mv2 split. Thanks to Graphcore IPU acceleration, GPS++ scales to deep architectures (16 layers), training at 3 minutes per epoch, and large ensemble (112 models), completing the final predictions in 1 hour 32 minutes, well under the 4 hour inference budget allocated. Our implementation is publicly available at: https://github.com/graphcore/ogb-lsc-pcqm4mv2.
Extreme Acceleration of Graph Neural Network-based Prediction Models for Quantum Chemistry
Nov 25, 2022Molecular property calculations are the bedrock of chemical physics. High-fidelity \textit{ab initio} modeling techniques for computing the molecular properties can be prohibitively expensive, and motivate the development of machine-learning models that make the same predictions more efficiently. Training graph neural networks over large molecular databases introduces unique computational challenges such as the need to process millions of small graphs with variable size and support communication patterns that are distinct from learning over large graphs such as social networks. This paper demonstrates a novel hardware-software co-design approach to scale up the training of graph neural networks for molecular property prediction. We introduce an algorithm to coalesce the batches of molecular graphs into fixed size packs to eliminate redundant computation and memory associated with alternative padding techniques and improve throughput via minimizing communication. We demonstrate the effectiveness of our co-design approach by providing an implementation of a well-established molecular property prediction model on the Graphcore Intelligence Processing Units (IPU). We evaluate the training performance on multiple molecular graph databases with varying degrees of graph counts, sizes and sparsity. We demonstrate that such a co-design approach can reduce the training time of such molecular property prediction models from days to less than two hours, opening new possibilities for AI-driven scientific discovery.
Reducing Down(stream)time: Pretraining Molecular GNNs using Heterogeneous AI Accelerators
Nov 08, 2022The demonstrated success of transfer learning has popularized approaches that involve pretraining models from massive data sources and subsequent finetuning towards a specific task. While such approaches have become the norm in fields such as natural language processing, implementation and evaluation of transfer learning approaches for chemistry are in the early stages. In this work, we demonstrate finetuning for downstream tasks on a graph neural network (GNN) trained over a molecular database containing 2.7 million water clusters. The use of Graphcore IPUs as an AI accelerator for training molecular GNNs reduces training time from a reported 2.7 days on 0.5M clusters to 1.2 hours on 2.7M clusters. Finetuning the pretrained model for downstream tasks of molecular dynamics and transfer to a different potential energy surface took only 8.3 hours and 28 minutes, respectively, on a single GPU.
Tuple Packing: Efficient Batching of Small Graphs in Graph Neural Networks
Sep 18, 2022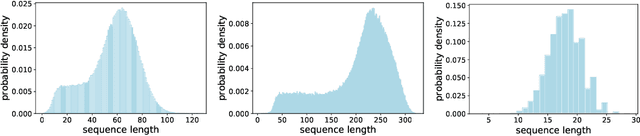
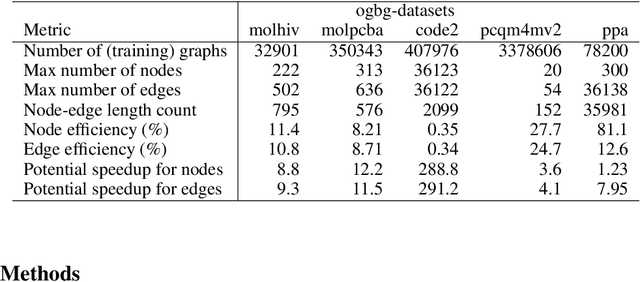
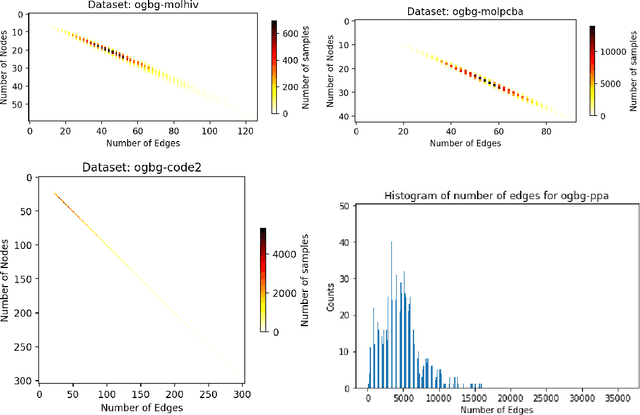
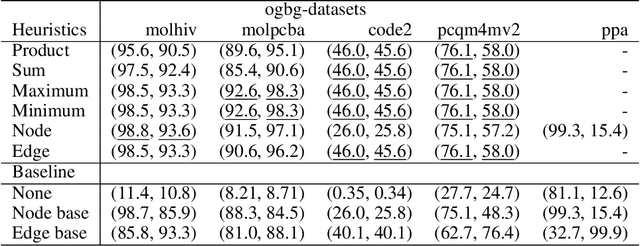
When processing a batch of graphs in machine learning models such as Graph Neural Networks (GNN), it is common to combine several small graphs into one overall graph to accelerate processing and remove or reduce the overhead of padding. This is for example supported in the PyG library. However, the sizes of small graphs can vary substantially with respect to the number of nodes and edges, and hence the size of the combined graph can still vary considerably, especially for small batch sizes. Therefore, the costs of excessive padding and wasted compute are still incurred when working with static shapes, which are preferred for maximum acceleration. This paper proposes a new hardware agnostic approach -- tuple packing -- for generating batches that cause minimal overhead. The algorithm extends recently introduced sequence packing approaches to work on the 2D tuples of (|nodes|, |edges|). A monotone heuristic is applied to the 2D histogram of tuple values to define a priority for packing histogram bins together with the objective to reach a limit on the number of nodes as well as the number of edges. Experiments verify the effectiveness of the algorithm on multiple datasets.