 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWeakly Supervised AI for Efficient Analysis of 3D Pathology Samples
Jul 27, 2023



Human tissue and its constituent cells form a microenvironment that is fundamentally three-dimensional (3D). However, the standard-of-care in pathologic diagnosis involves selecting a few two-dimensional (2D) sections for microscopic evaluation, risking sampling bias and misdiagnosis. Diverse methods for capturing 3D tissue morphologies have been developed, but they have yet had little translation to clinical practice; manual and computational evaluations of such large 3D data have so far been impractical and/or unable to provide patient-level clinical insights. Here we present Modality-Agnostic Multiple instance learning for volumetric Block Analysis (MAMBA), a deep-learning-based platform for processing 3D tissue images from diverse imaging modalities and predicting patient outcomes. Archived prostate cancer specimens were imaged with open-top light-sheet microscopy or microcomputed tomography and the resulting 3D datasets were used to train risk-stratification networks based on 5-year biochemical recurrence outcomes via MAMBA. With the 3D block-based approach, MAMBA achieves an area under the receiver operating characteristic curve (AUC) of 0.86 and 0.74, superior to 2D traditional single-slice-based prognostication (AUC of 0.79 and 0.57), suggesting superior prognostication with 3D morphological features. Further analyses reveal that the incorporation of greater tissue volume improves prognostic performance and mitigates risk prediction variability from sampling bias, suggesting the value of capturing larger extents of heterogeneous 3D morphology. With the rapid growth and adoption of 3D spatial biology and pathology techniques by researchers and clinicians, MAMBA provides a general and efficient framework for 3D weakly supervised learning for clinical decision support and can help to reveal novel 3D morphological biomarkers for prognosis and therapeutic response.
Towards a Visual-Language Foundation Model for Computational Pathology
Jul 25, 2023



The accelerated adoption of digital pathology and advances in deep learning have enabled the development of powerful models for various pathology tasks across a diverse array of diseases and patient cohorts. However, model training is often difficult due to label scarcity in the medical domain and the model's usage is limited by the specific task and disease for which it is trained. Additionally, most models in histopathology leverage only image data, a stark contrast to how humans teach each other and reason about histopathologic entities. We introduce CONtrastive learning from Captions for Histopathology (CONCH), a visual-language foundation model developed using diverse sources of histopathology images, biomedical text, and notably over 1.17 million image-caption pairs via task-agnostic pretraining. Evaluated on a suite of 13 diverse benchmarks, CONCH can be transferred to a wide range of downstream tasks involving either or both histopathology images and text, achieving state-of-the-art performance on histology image classification, segmentation, captioning, text-to-image and image-to-text retrieval. CONCH represents a substantial leap over concurrent visual-language pretrained systems for histopathology, with the potential to directly facilitate a wide array of machine learning-based workflows requiring minimal or no further supervised fine-tuning.
Visual Language Pretrained Multiple Instance Zero-Shot Transfer for Histopathology Images
Jun 13, 2023Contrastive visual language pretraining has emerged as a powerful method for either training new language-aware image encoders or augmenting existing pretrained models with zero-shot visual recognition capabilities. However, existing works typically train on large datasets of image-text pairs and have been designed to perform downstream tasks involving only small to medium sized-images, neither of which are applicable to the emerging field of computational pathology where there are limited publicly available paired image-text datasets and each image can span up to 100,000 x 100,000 pixels. In this paper we present MI-Zero, a simple and intuitive framework for unleashing the zero-shot transfer capabilities of contrastively aligned image and text models on gigapixel histopathology whole slide images, enabling multiple downstream diagnostic tasks to be carried out by pretrained encoders without requiring any additional labels. MI-Zero reformulates zero-shot transfer under the framework of multiple instance learning to overcome the computational challenge of inference on extremely large images. We used over 550k pathology reports and other available in-domain text corpora to pre-train our text encoder. By effectively leveraging strong pre-trained encoders, our best model pretrained on over 33k histopathology image-caption pairs achieves an average median zero-shot accuracy of 70.2% across three different real-world cancer subtyping tasks. Our code is available at: https://github.com/mahmoodlab/MI-Zero.
Incorporating intratumoral heterogeneity into weakly-supervised deep learning models via variance pooling
Jun 17, 2022



Supervised learning tasks such as cancer survival prediction from gigapixel whole slide images (WSIs) are a critical challenge in computational pathology that requires modeling complex features of the tumor microenvironment. These learning tasks are often solved with deep multi-instance learning (MIL) models that do not explicitly capture intratumoral heterogeneity. We develop a novel variance pooling architecture that enables a MIL model to incorporate intratumoral heterogeneity into its predictions. Two interpretability tools based on representative patches are illustrated to probe the biological signals captured by these models. An empirical study with 4,479 gigapixel WSIs from the Cancer Genome Atlas shows that adding variance pooling onto MIL frameworks improves survival prediction performance for five cancer types.
Differentiable Zooming for Multiple Instance Learning on Whole-Slide Images
Apr 30, 2022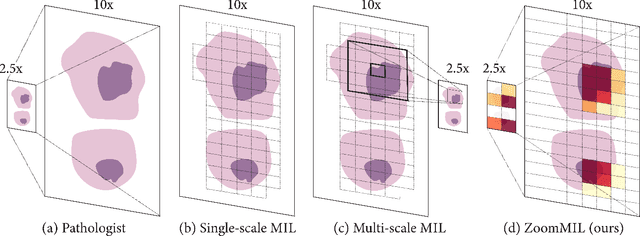
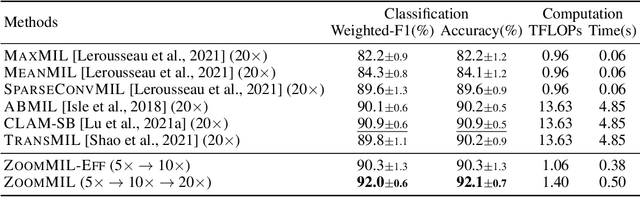
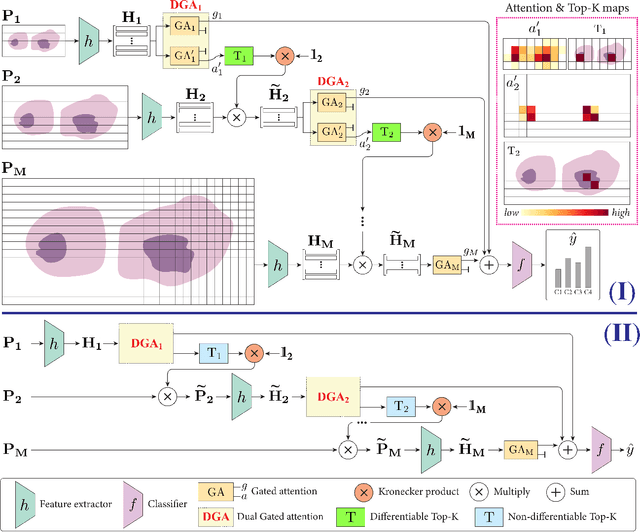
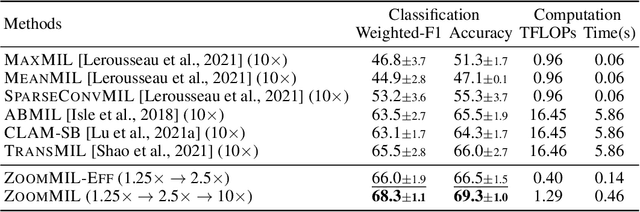
Multiple Instance Learning (MIL) methods have become increasingly popular for classifying giga-pixel sized Whole-Slide Images (WSIs) in digital pathology. Most MIL methods operate at a single WSI magnification, by processing all the tissue patches. Such a formulation induces high computational requirements, and constrains the contextualization of the WSI-level representation to a single scale. A few MIL methods extend to multiple scales, but are computationally more demanding. In this paper, inspired by the pathological diagnostic process, we propose ZoomMIL, a method that learns to perform multi-level zooming in an end-to-end manner. ZoomMIL builds WSI representations by aggregating tissue-context information from multiple magnifications. The proposed method outperforms the state-of-the-art MIL methods in WSI classification on two large datasets, while significantly reducing the computational demands with regard to Floating-Point Operations (FLOPs) and processing time by up to 40x.
Algorithm Fairness in AI for Medicine and Healthcare
Oct 01, 2021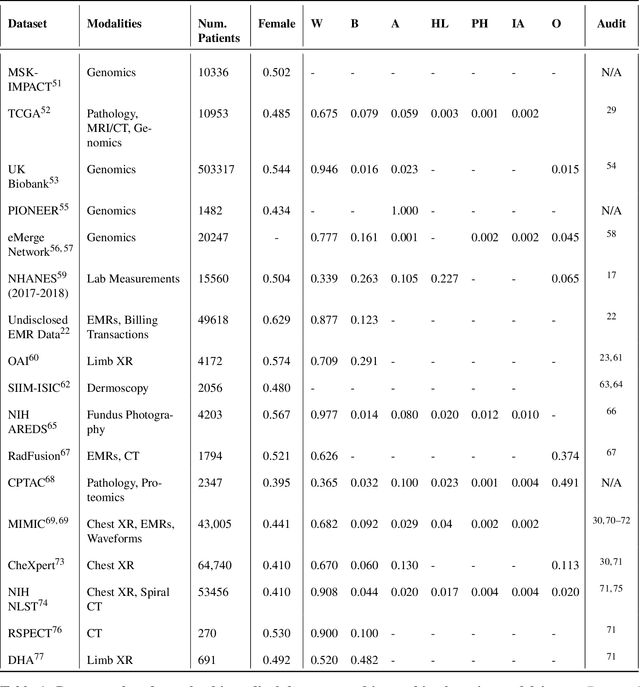
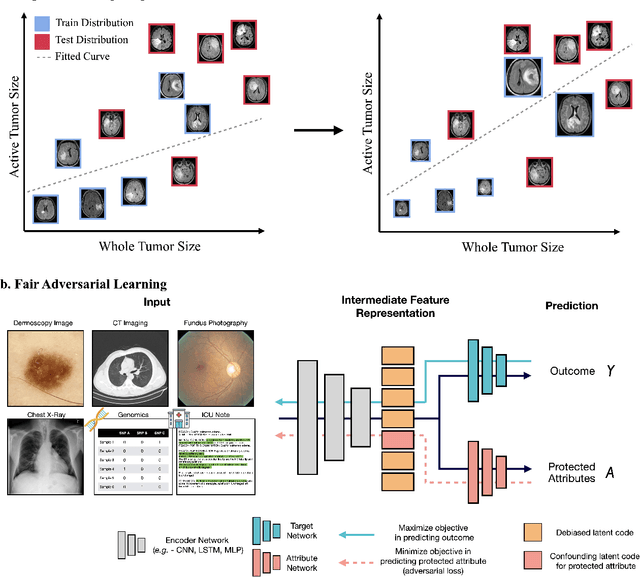
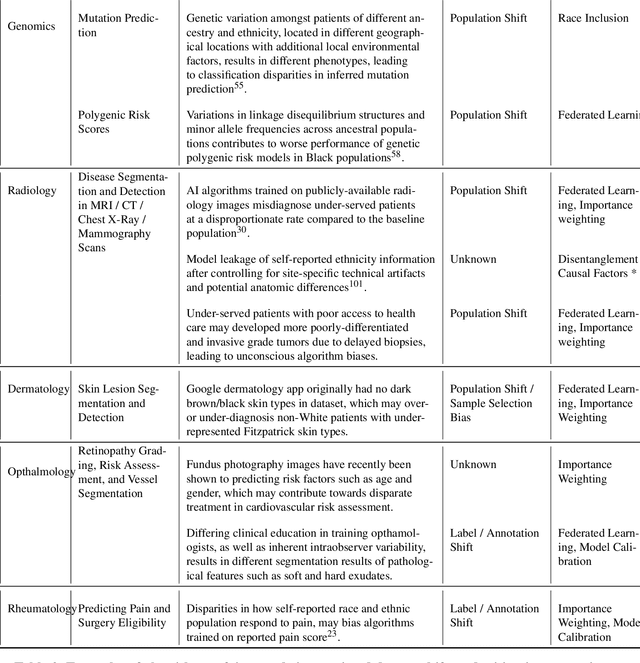
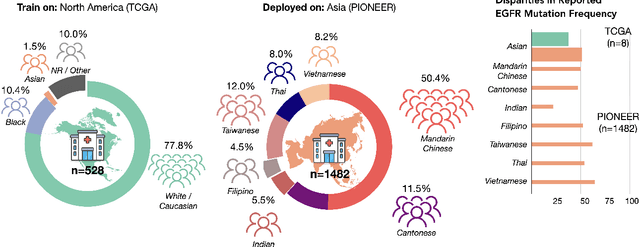
In the current development and deployment of many artificial intelligence (AI) systems in healthcare, algorithm fairness is a challenging problem in delivering equitable care. Recent evaluation of AI models stratified across race sub-populations have revealed enormous inequalities in how patients are diagnosed, given treatments, and billed for healthcare costs. In this perspective article, we summarize the intersectional field of fairness in machine learning through the context of current issues in healthcare, outline how algorithmic biases (e.g. - image acquisition, genetic variation, intra-observer labeling variability) arise in current clinical workflows and their resulting healthcare disparities. Lastly, we also review emerging strategies for mitigating bias via decentralized learning, disentanglement, and model explainability.
Pan-Cancer Integrative Histology-Genomic Analysis via Interpretable Multimodal Deep Learning
Aug 04, 2021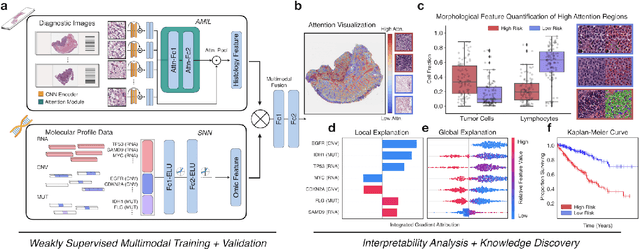
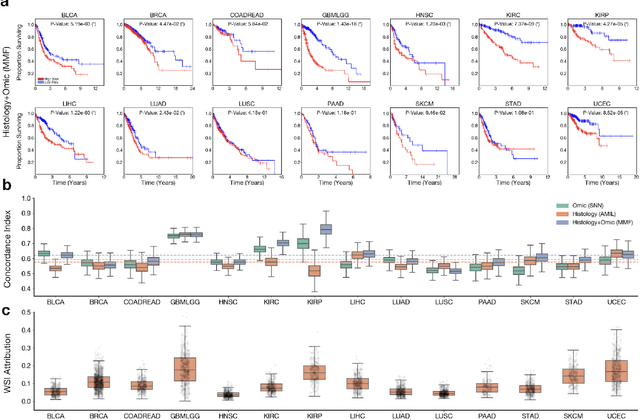
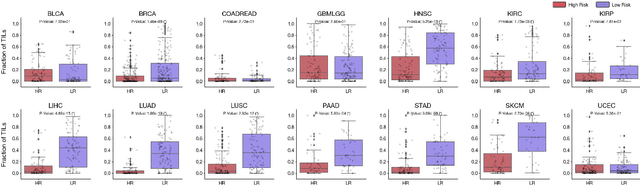
The rapidly emerging field of deep learning-based computational pathology has demonstrated promise in developing objective prognostic models from histology whole slide images. However, most prognostic models are either based on histology or genomics alone and do not address how histology and genomics can be integrated to develop joint image-omic prognostic models. Additionally identifying explainable morphological and molecular descriptors from these models that govern such prognosis is of interest. We used multimodal deep learning to integrate gigapixel whole slide pathology images, RNA-seq abundance, copy number variation, and mutation data from 5,720 patients across 14 major cancer types. Our interpretable, weakly-supervised, multimodal deep learning algorithm is able to fuse these heterogeneous modalities for predicting outcomes and discover prognostic features from these modalities that corroborate with poor and favorable outcomes via multimodal interpretability. We compared our model with unimodal deep learning models trained on histology slides and molecular profiles alone, and demonstrate performance increase in risk stratification on 9 out of 14 cancers. In addition, we analyze morphologic and molecular markers responsible for prognostic predictions across all cancer types. All analyzed data, including morphological and molecular correlates of patient prognosis across the 14 cancer types at a disease and patient level are presented in an interactive open-access database (http://pancancer.mahmoodlab.org) to allow for further exploration and prognostic biomarker discovery. To validate that these model explanations are prognostic, we further analyzed high attention morphological regions in WSIs, which indicates that tumor-infiltrating lymphocyte presence corroborates with favorable cancer prognosis on 9 out of 14 cancer types studied.
Fast and Scalable Image Search For Histology
Jul 28, 2021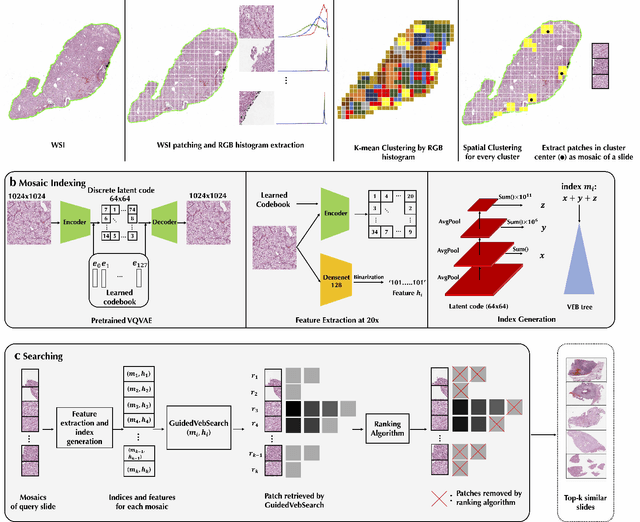
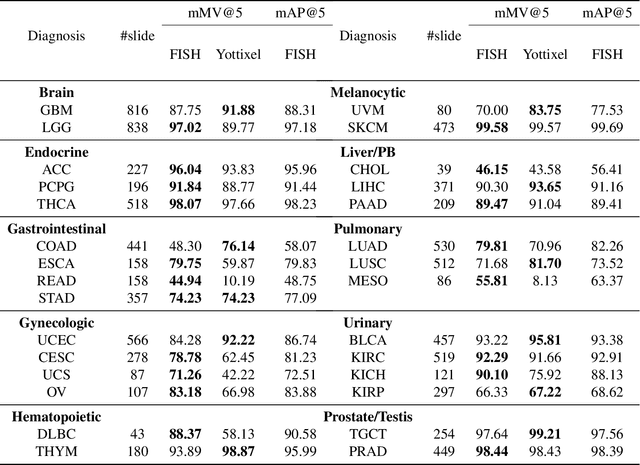
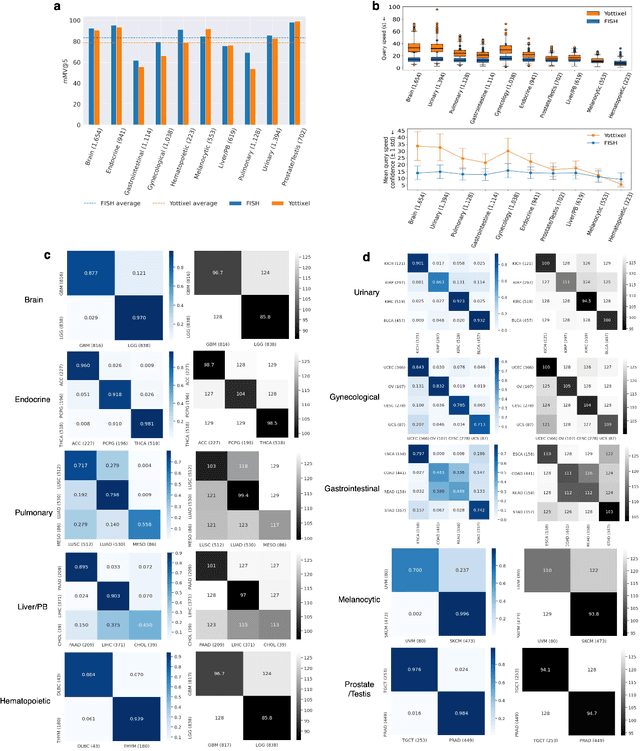
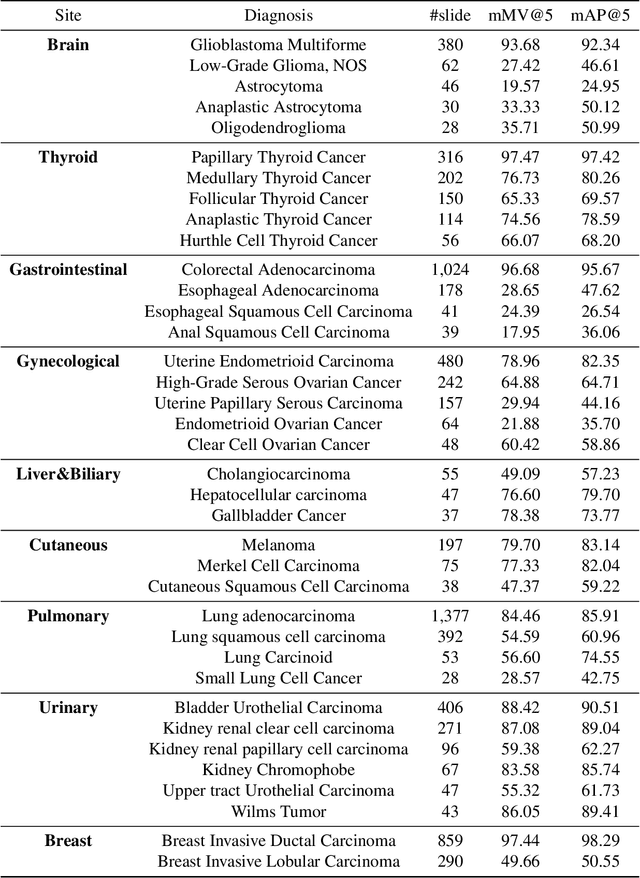
The expanding adoption of digital pathology has enabled the curation of large repositories of histology whole slide images (WSIs), which contain a wealth of information. Similar pathology image search offers the opportunity to comb through large historical repositories of gigapixel WSIs to identify cases with similar morphological features and can be particularly useful for diagnosing rare diseases, identifying similar cases for predicting prognosis, treatment outcomes, and potential clinical trial success. A critical challenge in developing a WSI search and retrieval system is scalability, which is uniquely challenging given the need to search a growing number of slides that each can consist of billions of pixels and are several gigabytes in size. Such systems are typically slow and retrieval speed often scales with the size of the repository they search through, making their clinical adoption tedious and are not feasible for repositories that are constantly growing. Here we present Fast Image Search for Histopathology (FISH), a histology image search pipeline that is infinitely scalable and achieves constant search speed that is independent of the image database size while being interpretable and without requiring detailed annotations. FISH uses self-supervised deep learning to encode meaningful representations from WSIs and a Van Emde Boas tree for fast search, followed by an uncertainty-based ranking algorithm to retrieve similar WSIs. We evaluated FISH on multiple tasks and datasets with over 22,000 patient cases spanning 56 disease subtypes. We additionally demonstrate that FISH can be used to assist with the diagnosis of rare cancer types where sufficient cases may not be available to train traditional supervised deep models. FISH is available as an easy-to-use, open-source software package (https://github.com/mahmoodlab/FISH).
Deep Learning-based Frozen Section to FFPE Translation
Jul 27, 2021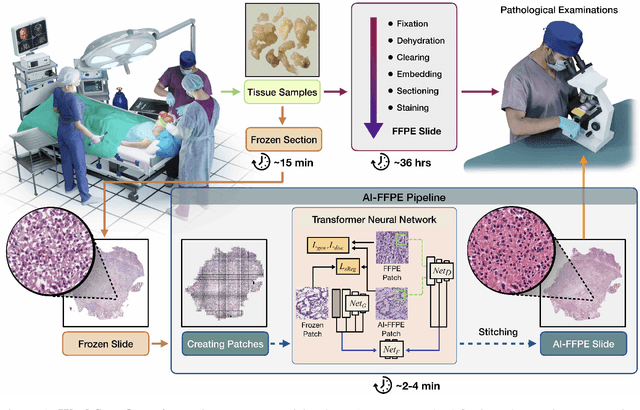
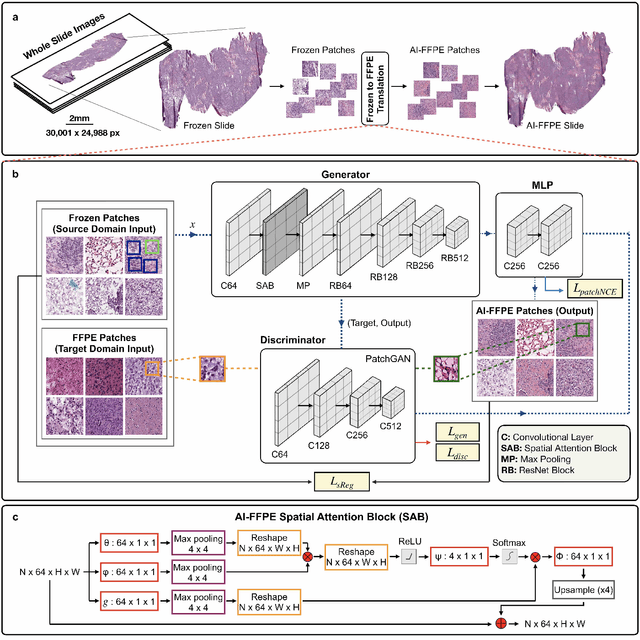
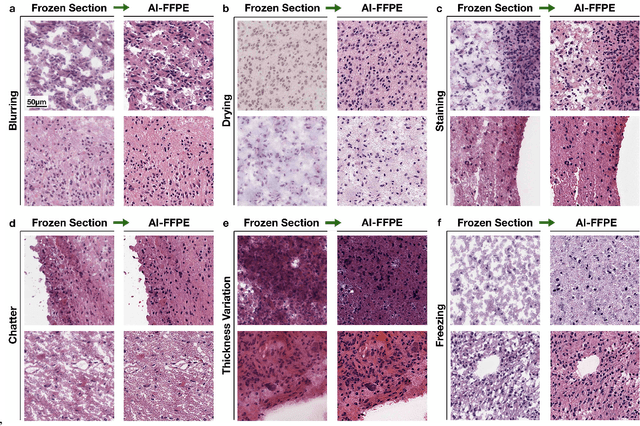
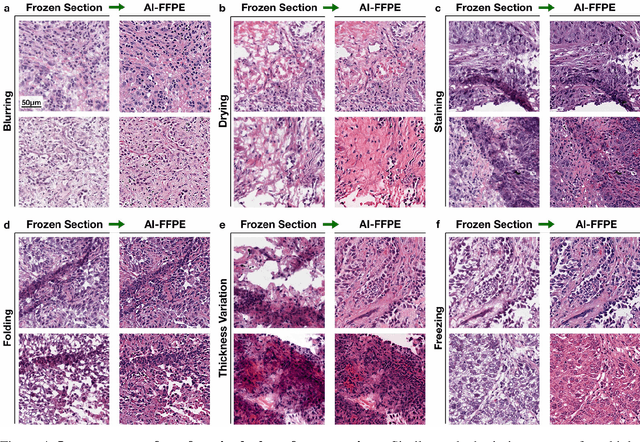
Frozen sectioning (FS) is the preparation method of choice for microscopic evaluation of tissues during surgical operations. The high speed of the procedure allows pathologists to rapidly assess the key microscopic features, such as tumour margins and malignant status to guide surgical decision-making and minimise disruptions to the course of the operation. However, FS is prone to introducing many misleading artificial structures (histological artefacts), such as nuclear ice crystals, compression, and cutting artefacts, hindering timely and accurate diagnostic judgement of the pathologist. Additional training and prolonged experience is often required to make highly effective and time-critical diagnosis on frozen sections. On the other hand, the gold standard tissue preparation technique of formalin-fixation and paraffin-embedding (FFPE) provides significantly superior image quality, but is a very time-consuming process (12-48 hours), making it unsuitable for intra-operative use. In this paper, we propose an artificial intelligence (AI) method that improves FS image quality by computationally transforming frozen-sectioned whole-slide images (FS-WSIs) into whole-slide FFPE-style images in minutes. AI-FFPE rectifies FS artefacts with the guidance of an attention mechanism that puts a particular emphasis on artefacts while utilising a self-regularization mechanism established between FS input image and synthesized FFPE-style image that preserves clinically relevant features. As a result, AI-FFPE method successfully generates FFPE-style images without significantly extending tissue processing time and consequently improves diagnostic accuracy. We demonstrate the efficacy of AI-FFPE on lung and brain frozen sections using a variety of different qualitative and quantitative metrics including visual Turing tests from 20 board certified pathologists.
Whole Slide Images are 2D Point Clouds: Context-Aware Survival Prediction using Patch-based Graph Convolutional Networks
Jul 27, 2021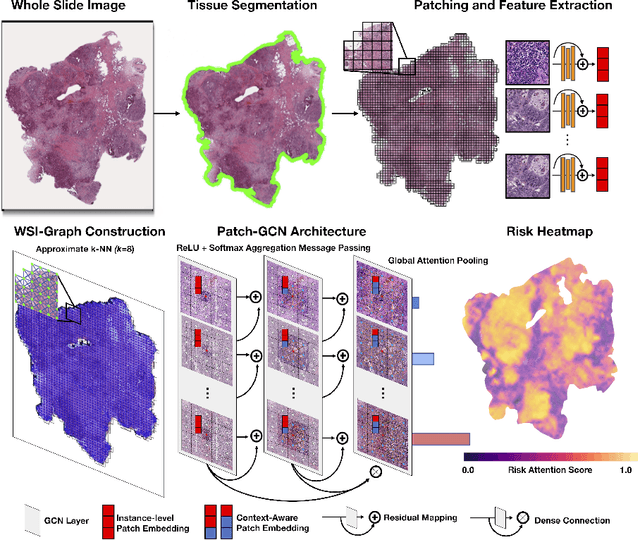
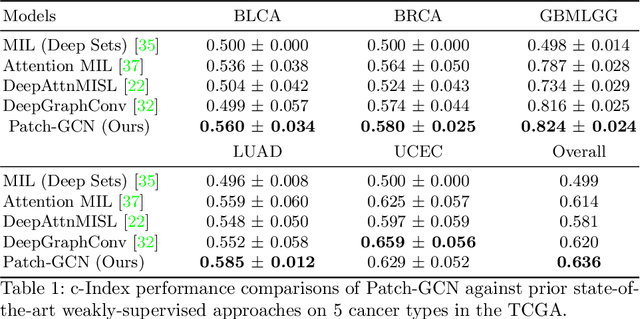
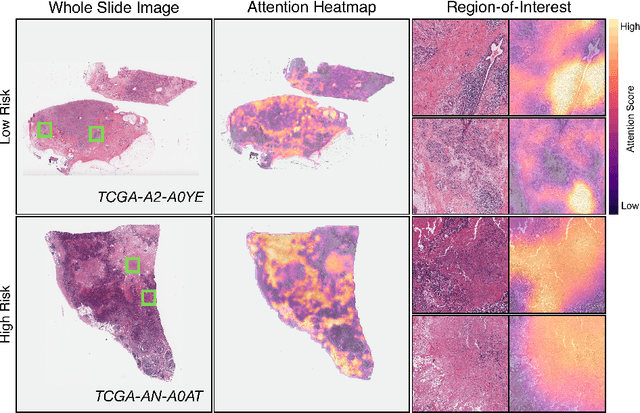
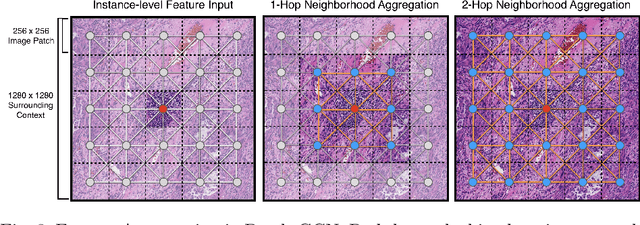
Cancer prognostication is a challenging task in computational pathology that requires context-aware representations of histology features to adequately infer patient survival. Despite the advancements made in weakly-supervised deep learning, many approaches are not context-aware and are unable to model important morphological feature interactions between cell identities and tissue types that are prognostic for patient survival. In this work, we present Patch-GCN, a context-aware, spatially-resolved patch-based graph convolutional network that hierarchically aggregates instance-level histology features to model local- and global-level topological structures in the tumor microenvironment. We validate Patch-GCN with 4,370 gigapixel WSIs across five different cancer types from the Cancer Genome Atlas (TCGA), and demonstrate that Patch-GCN outperforms all prior weakly-supervised approaches by 3.58-9.46%. Our code and corresponding models are publicly available at https://github.com/mahmoodlab/Patch-GCN.