 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSystem of Agentic AI for the Discovery of Metal-Organic Frameworks
Apr 18, 2025Generative models and machine learning promise accelerated material discovery in MOFs for CO2 capture and water harvesting but face significant challenges navigating vast chemical spaces while ensuring synthetizability. Here, we present MOFGen, a system of Agentic AI comprising interconnected agents: a large language model that proposes novel MOF compositions, a diffusion model that generates crystal structures, quantum mechanical agents that optimize and filter candidates, and synthetic-feasibility agents guided by expert rules and machine learning. Trained on all experimentally reported MOFs and computational databases, MOFGen generated hundreds of thousands of novel MOF structures and synthesizable organic linkers. Our methodology was validated through high-throughput experiments and the successful synthesis of five "AI-dreamt" MOFs, representing a major step toward automated synthesizable material discovery.
Manifold-Constrained Nucleus-Level Denoising Diffusion Model for Structure-Based Drug Design
Sep 16, 2024Artificial intelligence models have shown great potential in structure-based drug design, generating ligands with high binding affinities. However, existing models have often overlooked a crucial physical constraint: atoms must maintain a minimum pairwise distance to avoid separation violation, a phenomenon governed by the balance of attractive and repulsive forces. To mitigate such separation violations, we propose NucleusDiff. It models the interactions between atomic nuclei and their surrounding electron clouds by enforcing the distance constraint between the nuclei and manifolds. We quantitatively evaluate NucleusDiff using the CrossDocked2020 dataset and a COVID-19 therapeutic target, demonstrating that NucleusDiff reduces violation rate by up to 100.00% and enhances binding affinity by up to 22.16%, surpassing state-of-the-art models for structure-based drug design. We also provide qualitative analysis through manifold sampling, visually confirming the effectiveness of NucleusDiff in reducing separation violations and improving binding affinities.
Single and Multi-Hop Question-Answering Datasets for Reticular Chemistry with GPT-4-Turbo
May 03, 2024The rapid advancement in artificial intelligence and natural language processing has led to the development of large-scale datasets aimed at benchmarking the performance of machine learning models. Herein, we introduce 'RetChemQA,' a comprehensive benchmark dataset designed to evaluate the capabilities of such models in the domain of reticular chemistry. This dataset includes both single-hop and multi-hop question-answer pairs, encompassing approximately 45,000 Q&As for each type. The questions have been extracted from an extensive corpus of literature containing about 2,530 research papers from publishers including NAS, ACS, RSC, Elsevier, and Nature Publishing Group, among others. The dataset has been generated using OpenAI's GPT-4 Turbo, a cutting-edge model known for its exceptional language understanding and generation capabilities. In addition to the Q&A dataset, we also release a dataset of synthesis conditions extracted from the corpus of literature used in this study. The aim of RetChemQA is to provide a robust platform for the development and evaluation of advanced machine learning algorithms, particularly for the reticular chemistry community. The dataset is structured to reflect the complexities and nuances of real-world scientific discourse, thereby enabling nuanced performance assessments across a variety of tasks. The dataset is available at the following link: https://github.com/nakulrampal/RetChemQA
A Multi-Grained Symmetric Differential Equation Model for Learning Protein-Ligand Binding Dynamics
Feb 01, 2024In drug discovery, molecular dynamics (MD) simulation for protein-ligand binding provides a powerful tool for predicting binding affinities, estimating transport properties, and exploring pocket sites. There has been a long history of improving the efficiency of MD simulations through better numerical methods and, more recently, by utilizing machine learning (ML) methods. Yet, challenges remain, such as accurate modeling of extended-timescale simulations. To address this issue, we propose NeuralMD, the first ML surrogate that can facilitate numerical MD and provide accurate simulations in protein-ligand binding. We propose a principled approach that incorporates a novel physics-informed multi-grained group symmetric framework. Specifically, we propose (1) a BindingNet model that satisfies group symmetry using vector frames and captures the multi-level protein-ligand interactions, and (2) an augmented neural differential equation solver that learns the trajectory under Newtonian mechanics. For the experiment, we design ten single-trajectory and three multi-trajectory binding simulation tasks. We show the efficiency and effectiveness of NeuralMD, with a 2000$\times$ speedup over standard numerical MD simulation and outperforming all other ML approaches by up to 80% under the stability metric. We further qualitatively show that NeuralMD reaches more stable binding predictions compared to other machine learning methods.
Image and Data Mining in Reticular Chemistry Using GPT-4V
Dec 09, 2023The integration of artificial intelligence into scientific research has reached a new pinnacle with GPT-4V, a large language model featuring enhanced vision capabilities, accessible through ChatGPT or an API. This study demonstrates the remarkable ability of GPT-4V to navigate and obtain complex data for metal-organic frameworks, especially from graphical sources. Our approach involved an automated process of converting 346 scholarly articles into 6240 images, which represents a benchmark dataset in this task, followed by deploying GPT-4V to categorize and analyze these images using natural language prompts. This methodology enabled GPT-4V to accurately identify and interpret key plots integral to MOF characterization, such as nitrogen isotherms, PXRD patterns, and TGA curves, among others, with accuracy and recall above 93%. The model's proficiency in extracting critical information from these plots not only underscores its capability in data mining but also highlights its potential in aiding the creation of comprehensive digital databases for reticular chemistry. In addition, the extracted nitrogen isotherm data from the selected literature allowed for a comparison between theoretical and experimental porosity values for over 200 compounds, highlighting certain discrepancies and underscoring the importance of integrating computational and experimental data. This work highlights the potential of AI in accelerating scientific discovery and innovation, bridging the gap between computational tools and experimental research, and paving the way for more efficient, inclusive, and comprehensive scientific inquiry.
GPT-4 Reticular Chemist for MOF Discovery
Jun 20, 2023



We present a new framework integrating the AI model GPT-4 into the iterative process of reticular chemistry experimentation, leveraging a cooperative workflow of interaction between AI and a human apprentice. This GPT-4 Reticular Chemist is an integrated system composed of three phases. Each of these utilizes GPT-4 in various capacities, wherein GPT-4 provides detailed instructions for chemical experimentation and the apprentice provides feedback on the experimental outcomes, including both success and failures, for the in-text learning of AI in the next iteration. This iterative human-AI interaction enabled GPT-4 to learn from the outcomes, much like an experienced chemist, by a prompt-learning strategy. Importantly, the system is based on natural language for both development and operation, eliminating the need for coding skills, and thus, make it accessible to all chemists. Our GPT-4 Reticular Chemist demonstrated the discovery of an isoreticular series of metal-organic frameworks (MOFs), each of which was made using distinct synthesis strategies and optimal conditions. This workflow presents a potential for broader applications in scientific research by harnessing the capability of large language models like GPT-4 to enhance the feasibility and efficiency of research activities.
ChatGPT Chemistry Assistant for Text Mining and Prediction of MOF Synthesis
Jun 20, 2023



We use prompt engineering to guide ChatGPT in the automation of text mining of metal-organic frameworks (MOFs) synthesis conditions from diverse formats and styles of the scientific literature. This effectively mitigates ChatGPT's tendency to hallucinate information -- an issue that previously made the use of Large Language Models (LLMs) in scientific fields challenging. Our approach involves the development of a workflow implementing three different processes for text mining, programmed by ChatGPT itself. All of them enable parsing, searching, filtering, classification, summarization, and data unification with different tradeoffs between labor, speed, and accuracy. We deploy this system to extract 26,257 distinct synthesis parameters pertaining to approximately 800 MOFs sourced from peer-reviewed research articles. This process incorporates our ChemPrompt Engineering strategy to instruct ChatGPT in text mining, resulting in impressive precision, recall, and F1 scores of 90-99%. Furthermore, with the dataset built by text mining, we constructed a machine-learning model with over 86% accuracy in predicting MOF experimental crystallization outcomes and preliminarily identifying important factors in MOF crystallization. We also developed a reliable data-grounded MOF chatbot to answer questions on chemical reactions and synthesis procedures. Given that the process of using ChatGPT reliably mines and tabulates diverse MOF synthesis information in a unified format, while using only narrative language requiring no coding expertise, we anticipate that our ChatGPT Chemistry Assistant will be very useful across various other chemistry sub-disciplines.
Symmetry-Informed Geometric Representation for Molecules, Proteins, and Crystalline Materials
Jun 15, 2023Artificial intelligence for scientific discovery has recently generated significant interest within the machine learning and scientific communities, particularly in the domains of chemistry, biology, and material discovery. For these scientific problems, molecules serve as the fundamental building blocks, and machine learning has emerged as a highly effective and powerful tool for modeling their geometric structures. Nevertheless, due to the rapidly evolving process of the field and the knowledge gap between science (e.g., physics, chemistry, & biology) and machine learning communities, a benchmarking study on geometrical representation for such data has not been conducted. To address such an issue, in this paper, we first provide a unified view of the current symmetry-informed geometric methods, classifying them into three main categories: invariance, equivariance with spherical frame basis, and equivariance with vector frame basis. Then we propose a platform, coined Geom3D, which enables benchmarking the effectiveness of geometric strategies. Geom3D contains 16 advanced symmetry-informed geometric representation models and 14 geometric pretraining methods over 46 diverse datasets, including small molecules, proteins, and crystalline materials. We hope that Geom3D can, on the one hand, eliminate barriers for machine learning researchers interested in exploring scientific problems; and, on the other hand, provide valuable guidance for researchers in computational chemistry, structural biology, and materials science, aiding in the informed selection of representation techniques for specific applications.
Disincentivizing Polarization in Social Networks
May 23, 2023



On social networks, algorithmic personalization drives users into filter bubbles where they rarely see content that deviates from their interests. We present a model for content curation and personalization that avoids filter bubbles, along with algorithmic guarantees and nearly matching lower bounds. In our model, the platform interacts with $n$ users over $T$ timesteps, choosing content for each user from $k$ categories. The platform receives stochastic rewards as in a multi-arm bandit. To avoid filter bubbles, we draw on the intuition that if some users are shown some category of content, then all users should see at least a small amount of that content. We first analyze a naive formalization of this intuition and show it has unintended consequences: it leads to ``tyranny of the majority'' with the burden of diversification borne disproportionately by those with minority interests. This leads us to our model which distributes this burden more equitably. We require that the probability any user is shown a particular type of content is at least $\gamma$ times the average probability all users are shown that type of content. Full personalization corresponds to $\gamma = 0$ and complete homogenization corresponds to $\gamma = 1$; hence, $\gamma$ encodes a hard cap on the level of personalization. We also analyze additional formulations where the platform can exceed its cap but pays a penalty proportional to its constraint violation. We provide algorithmic guarantees for optimizing recommendations subject to these constraints. These include nearly matching upper and lower bounds for the entire range of $\gamma \in [0,1]$ showing that the reward of a multi-agent variant of UCB is nearly optimal. Using real-world preference data, we empirically verify that under our model, users share the burden of diversification with only minor utility loss under our constraints.
Strategic Ranking
Sep 16, 2021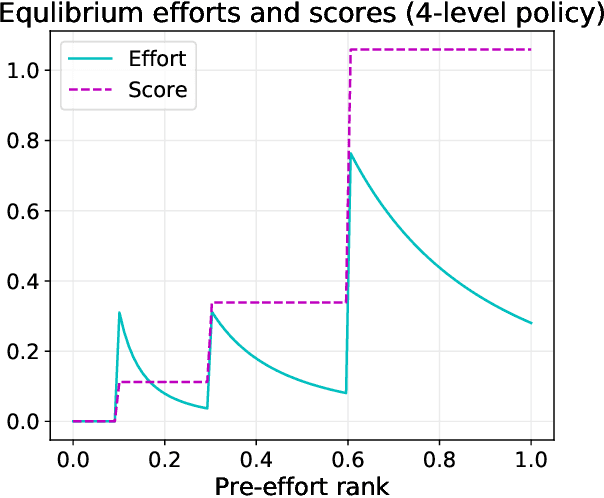

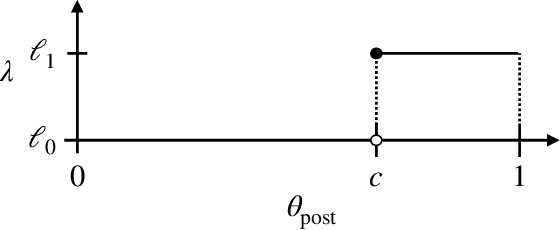
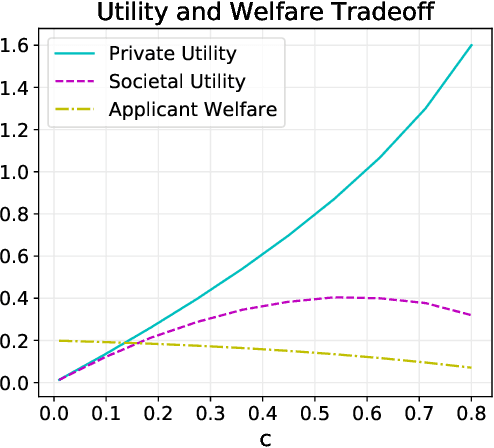
Strategic classification studies the design of a classifier robust to the manipulation of input by strategic individuals. However, the existing literature does not consider the effect of competition among individuals as induced by the algorithm design. Motivated by constrained allocation settings such as college admissions, we introduce strategic ranking, in which the (designed) individual reward depends on an applicant's post-effort rank in a measurement of interest. Our results illustrate how competition among applicants affects the resulting equilibria and model insights. We analyze how various ranking reward designs trade off applicant, school, and societal utility and in particular how ranking design can counter inequities arising from disparate access to resources to improve one's measured score: We find that randomization in the ranking reward design can mitigate two measures of disparate impact, welfare gap and access, whereas non-randomization may induce a high level of competition that systematically excludes a disadvantaged group.