 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeWhat makes for good morphology representations for spatial omics?
Aug 01, 2024



Spatial omics has transformed our understanding of tissue architecture by preserving spatial context of gene expression patterns. Simultaneously, advances in imaging AI have enabled extraction of morphological features describing the tissue. The intersection of spatial omics and imaging AI presents opportunities for a more holistic understanding. In this review we introduce a framework for categorizing spatial omics-morphology combination methods, focusing on how morphological features can be translated or integrated into spatial omics analyses. By translation we mean finding morphological features that spatially correlate with gene expression patterns with the purpose of predicting gene expression. Such features can be used to generate super-resolution gene expression maps or infer genetic information from clinical H&E-stained samples. By integration we mean finding morphological features that spatially complement gene expression patterns with the purpose of enriching information. Such features can be used to define spatial domains, especially where gene expression has preceded morphological changes and where morphology remains after gene expression. We discuss learning strategies and directions for further development of the field.
Transcriptome-supervised classification of tissue morphology using deep learning
Dec 07, 2023



Deep learning has proven to successfully learn variations in tissue and cell morphology. Training of such models typically relies on expensive manual annotations. Here we conjecture that spatially resolved gene expression, e.i., the transcriptome, can be used as an alternative to manual annotations. In particular, we trained five convolutional neural networks with patches of different size extracted from locations defined by spatially resolved gene expression. The network is trained to classify tissue morphology related to two different genes, general tissue, as well as background, on an image of fluorescence stained nuclei in a mouse brain coronal section. Performance is evaluated on an independent tissue section from a different mouse brain, reaching an average Dice score of 0.51. Results may indicate that novel techniques for spatially resolved transcriptomics together with deep learning may provide a unique and unbiased way to find genotype-phenotype relationships.
* Accepted for publication at IEEE International Symposium on Biomedical Imaging (ISBI) 2020
Roadmap on Deep Learning for Microscopy
Mar 07, 2023



Through digital imaging, microscopy has evolved from primarily being a means for visual observation of life at the micro- and nano-scale, to a quantitative tool with ever-increasing resolution and throughput. Artificial intelligence, deep neural networks, and machine learning are all niche terms describing computational methods that have gained a pivotal role in microscopy-based research over the past decade. This Roadmap is written collectively by prominent researchers and encompasses selected aspects of how machine learning is applied to microscopy image data, with the aim of gaining scientific knowledge by improved image quality, automated detection, segmentation, classification and tracking of objects, and efficient merging of information from multiple imaging modalities. We aim to give the reader an overview of the key developments and an understanding of possibilities and limitations of machine learning for microscopy. It will be of interest to a wide cross-disciplinary audience in the physical sciences and life sciences.
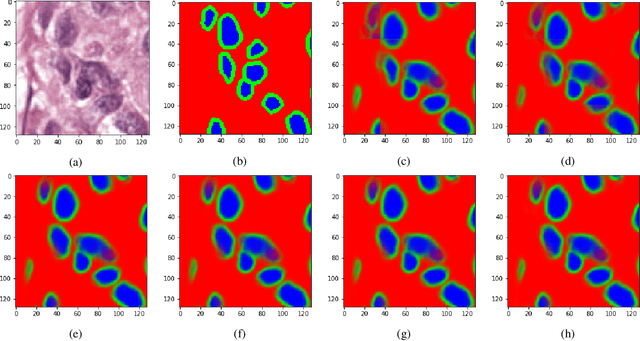
Seeded iterative clustering for histology region identification
Nov 14, 2022


Annotations are necessary to develop computer vision algorithms for histopathology, but dense annotations at a high resolution are often time-consuming to make. Deep learning models for segmentation are a way to alleviate the process, but require large amounts of training data, training times and computing power. To address these issues, we present seeded iterative clustering to produce a coarse segmentation densely and at the whole slide level. The algorithm uses precomputed representations as the clustering space and a limited amount of sparse interactive annotations as seeds to iteratively classify image patches. We obtain a fast and effective way of generating dense annotations for whole slide images and a framework that allows the comparison of neural network latent representations in the context of transfer learning.
CoMIR: Contrastive Multimodal Image Representation for Registration
Jun 11, 2020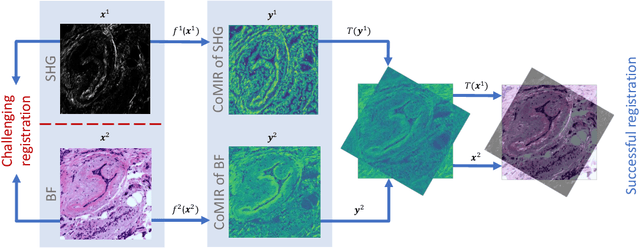

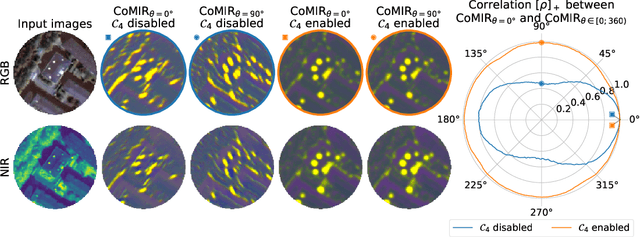
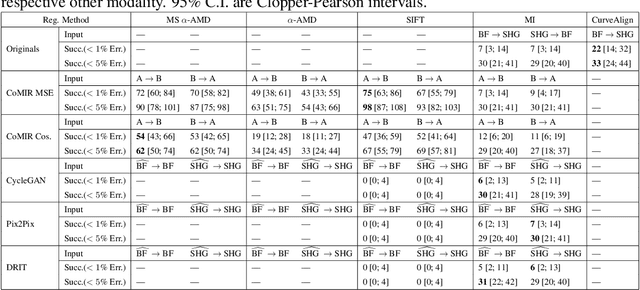
We propose contrastive coding to learn shared, dense image representations, referred to as CoMIRs (Contrastive Multimodal Image Representations). CoMIRs enable the registration of multimodal images where existing registration methods often fail due to a lack of sufficiently similar image structures. CoMIRs reduce the multimodal registration problem to a monomodal one in which general intensity-based, as well as feature-based, registration algorithms can be applied. The method involves training one neural network per modality on aligned images, using a contrastive loss based on noise-contrastive estimation (InfoNCE). Unlike other contrastive coding methods, used for e.g. classification, our approach generates image-like representations that contain the information shared between modalities. We introduce a novel, hyperparameter-free modification to InfoNCE, to enforce rotational equivariance of the learnt representations, a property essential to the registration task. We assess the extent of achieved rotational equivariance and the stability of the representations with respect to weight initialization, training set, and hyperparameter settings, on a remote sensing dataset of RGB and near-infrared images. We evaluate the learnt representations through registration of a biomedical dataset of bright-field and second-harmonic generation microscopy images; two modalities with very little apparent correlation. The proposed approach based on CoMIRs significantly outperforms registration of representations created by GAN-based image-to-image translation, as well as a state-of-the-art, application-specific method which takes additional knowledge about the data into account. Code is available at: https://github.com/dqiamsdoayehccdvulyy/CoMIR.
Introducing Hann windows for reducing edge-effects in patch-based image segmentation
Oct 17, 2019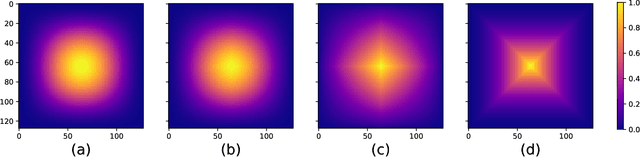
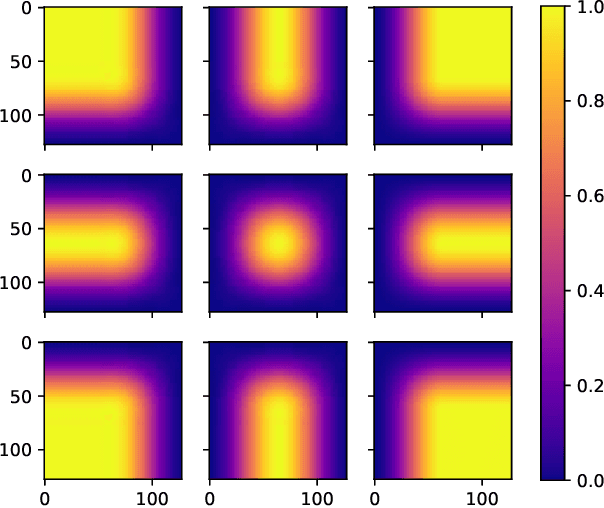
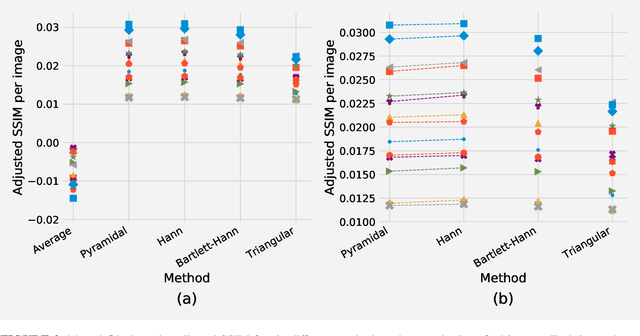
There is a limitation in the size of an image that can be processed using computationally demanding methods such as e.g. Convolutional Neural Networks (CNNs). Some imaging modalities - notably biological and medical - can result in images up to a few gigapixels in size, meaning that they have to be divided into smaller parts, or patches, for processing. However, when performing image segmentation, this may lead to undesirable artefacts, such as edge effects in the final re-combined image. We introduce windowing methods from signal processing to effectively reduce such edge effects. With the assumption that the central part of an image patch often holds richer contextual information than its sides and corners, we reconstruct the prediction by overlapping patches that are being weighted depending on 2-dimensional windows. We compare the results of four different windows: Hann, Bartlett-Hann, Triangular and a recently proposed window by Cui et al., and show that the cosine-based Hann window achieves the best improvement as measured by the Structural Similarity Index (SSIM). The proposed windowing method can be used together with any CNN model for segmentation without any modification and significantly improves network predictions.
Pathologist-Level Grading of Prostate Biopsies with Artificial Intelligence
Jul 02, 2019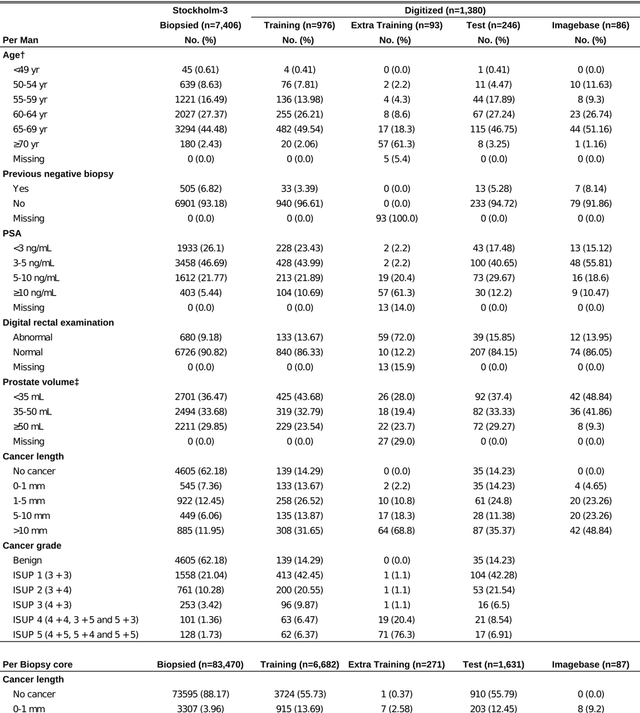
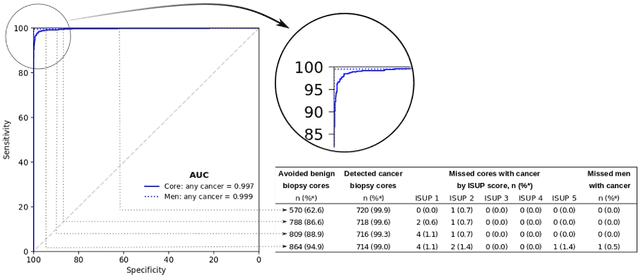
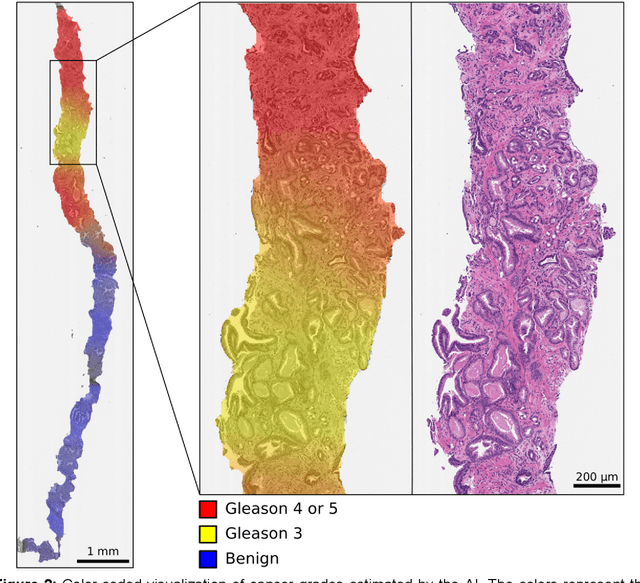
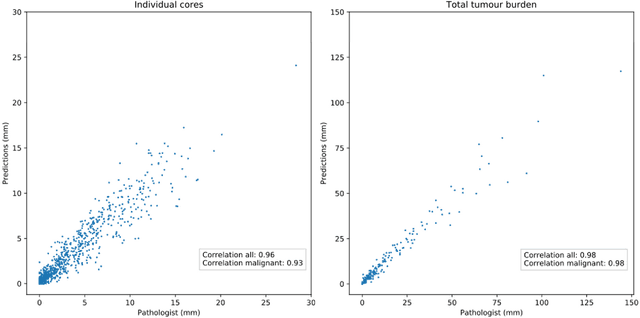
Background: An increasing volume of prostate biopsies and a world-wide shortage of uro-pathologists puts a strain on pathology departments. Additionally, the high intra- and inter-observer variability in grading can result in over- and undertreatment of prostate cancer. Artificial intelligence (AI) methods may alleviate these problems by assisting pathologists to reduce workload and harmonize grading. Methods: We digitized 6,682 needle biopsies from 976 participants in the population based STHLM3 diagnostic study to train deep neural networks for assessing prostate biopsies. The networks were evaluated by predicting the presence, extent, and Gleason grade of malignant tissue for an independent test set comprising 1,631 biopsies from 245 men. We additionally evaluated grading performance on 87 biopsies individually graded by 23 experienced urological pathologists from the International Society of Urological Pathology. We assessed discriminatory performance by receiver operating characteristics (ROC) and tumor extent predictions by correlating predicted millimeter cancer length against measurements by the reporting pathologist. We quantified the concordance between grades assigned by the AI and the expert urological pathologists using Cohen's kappa. Results: The performance of the AI to detect and grade cancer in prostate needle biopsy samples was comparable to that of international experts in prostate pathology. The AI achieved an area under the ROC curve of 0.997 for distinguishing between benign and malignant biopsy cores, and 0.999 for distinguishing between men with or without prostate cancer. The correlation between millimeter cancer predicted by the AI and assigned by the reporting pathologist was 0.96. For assigning Gleason grades, the AI achieved an average pairwise kappa of 0.62. This was within the range of the corresponding values for the expert pathologists (0.60 to 0.73).
Whole slide image registration for the study of tumor heterogeneity
Jan 24, 2019
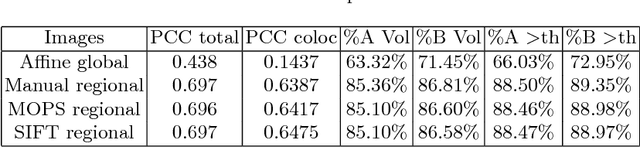
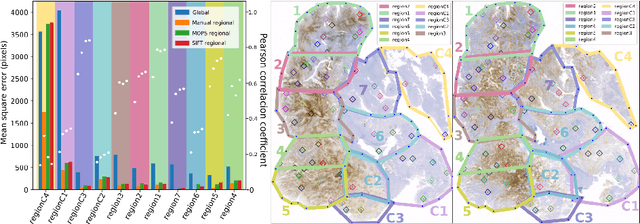
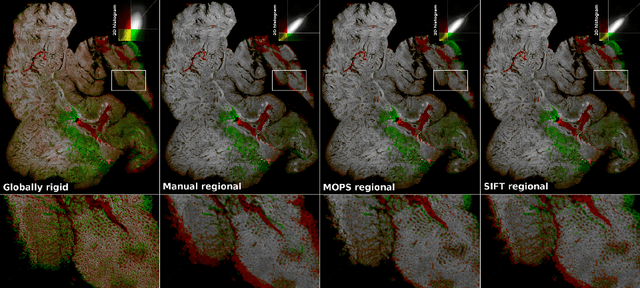
Consecutive thin sections of tissue samples make it possible to study local variation in e.g. protein expression and tumor heterogeneity by staining for a new protein in each section. In order to compare and correlate patterns of different proteins, the images have to be registered with high accuracy. The problem we want to solve is registration of gigapixel whole slide images (WSI). This presents 3 challenges: (i) Images are very large; (ii) Thin sections result in artifacts that make global affine registration prone to very large local errors; (iii) Local affine registration is required to preserve correct tissue morphology (local size, shape and texture). In our approach we compare WSI registration based on automatic and manual feature selection on either the full image or natural sub-regions (as opposed to square tiles). Working with natural sub-regions, in an interactive tool makes it possible to exclude regions containing scientifically irrelevant information. We also present a new way to visualize local registration quality by a Registration Confidence Map (RCM). With this method, intra-tumor heterogeneity and charateristics of the tumor microenvironment can be observed and quantified.
* MICCAI2018 - Computational Pathology and Ophthalmic Medical Image Analysis - COMPAY
Improving Recall of In Situ Sequencing by Self-Learned Features and a Graphical Model
Feb 24, 2018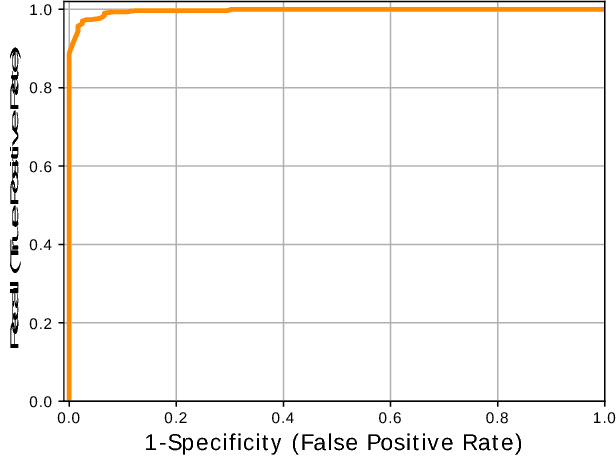
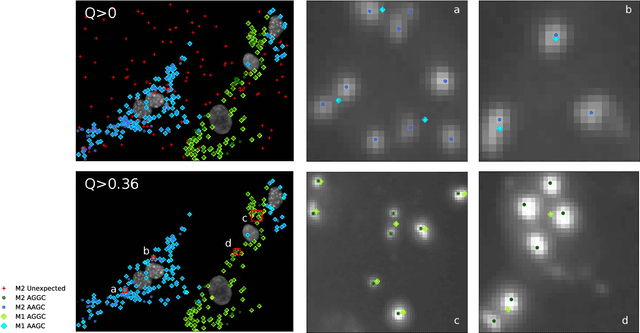
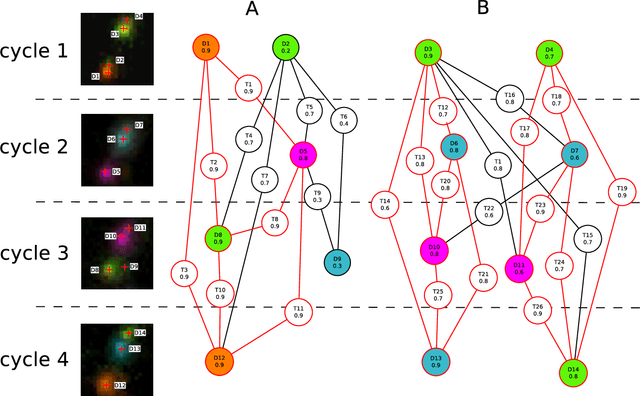
Image-based sequencing of mRNA makes it possible to see where in a tissue sample a given gene is active, and thus discern large numbers of different cell types in parallel. This is crucial for gaining a better understanding of tissue development and disease such as cancer. Signals are collected over multiple staining and imaging cycles, and signal density together with noise makes signal decoding challenging. Previous approaches have led to low signal recall in efforts to maintain high sensitivity. We propose an approach where signal candidates are generously included, and true-signal probability at the cycle level is self-learned using a convolutional neural network. Signal candidates and probability predictions are thereafter fed into a graphical model searching for signal candidates across sequencing cycles. The graphical model combines intensity, probability and spatial distance to find optimal paths representing decoded signal sequences. We evaluate our approach in relation to state-of-the-art, and show that we increase recall by $27\%$ at maintained sensitivity. Furthermore, visual examination shows that most of the now correctly resolved signals were previously lost due to high signal density. Thus, the proposed approach has the potential to significantly improve further analysis of spatial statistics in in situ sequencing experiments.