 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNavigating protein landscapes with a machine-learned transferable coarse-grained model
Oct 27, 2023



The most popular and universally predictive protein simulation models employ all-atom molecular dynamics (MD), but they come at extreme computational cost. The development of a universal, computationally efficient coarse-grained (CG) model with similar prediction performance has been a long-standing challenge. By combining recent deep learning methods with a large and diverse training set of all-atom protein simulations, we here develop a bottom-up CG force field with chemical transferability, which can be used for extrapolative molecular dynamics on new sequences not used during model parametrization. We demonstrate that the model successfully predicts folded structures, intermediates, metastable folded and unfolded basins, and the fluctuations of intrinsically disordered proteins while it is several orders of magnitude faster than an all-atom model. This showcases the feasibility of a universal and computationally efficient machine-learned CG model for proteins.
Machine Learning Coarse-Grained Potentials of Protein Thermodynamics
Dec 14, 2022A generalized understanding of protein dynamics is an unsolved scientific problem, the solution of which is critical to the interpretation of the structure-function relationships that govern essential biological processes. Here, we approach this problem by constructing coarse-grained molecular potentials based on artificial neural networks and grounded in statistical mechanics. For training, we build a unique dataset of unbiased all-atom molecular dynamics simulations of approximately 9 ms for twelve different proteins with multiple secondary structure arrangements. The coarse-grained models are capable of accelerating the dynamics by more than three orders of magnitude while preserving the thermodynamics of the systems. Coarse-grained simulations identify relevant structural states in the ensemble with comparable energetics to the all-atom systems. Furthermore, we show that a single coarse-grained potential can integrate all twelve proteins and can capture experimental structural features of mutated proteins. These results indicate that machine learning coarse-grained potentials could provide a feasible approach to simulate and understand protein dynamics.
Deeptime: a Python library for machine learning dynamical models from time series data
Oct 28, 2021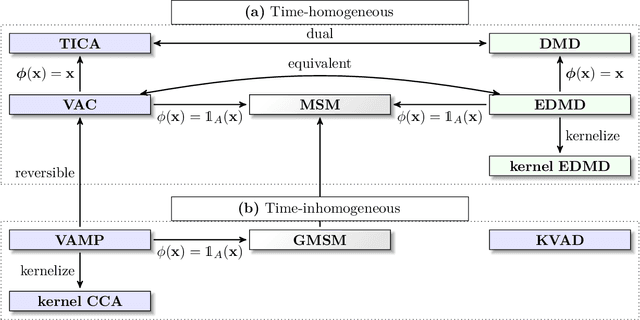

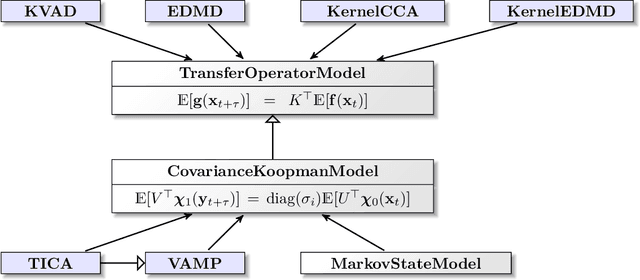
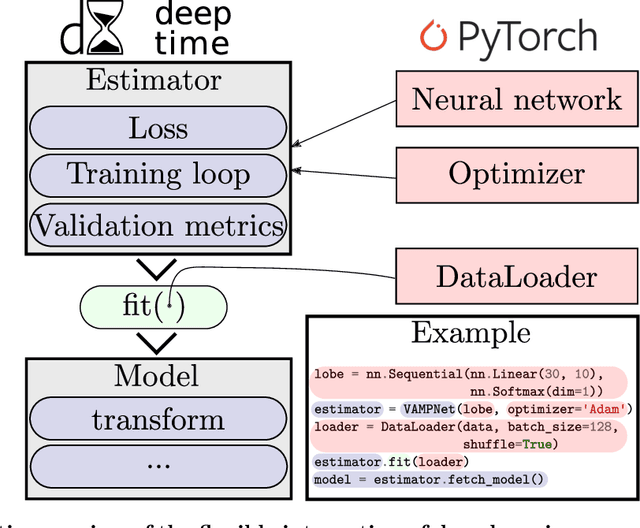
Generation and analysis of time-series data is relevant to many quantitative fields ranging from economics to fluid mechanics. In the physical sciences, structures such as metastable and coherent sets, slow relaxation processes, collective variables dominant transition pathways or manifolds and channels of probability flow can be of great importance for understanding and characterizing the kinetic, thermodynamic and mechanistic properties of the system. Deeptime is a general purpose Python library offering various tools to estimate dynamical models based on time-series data including conventional linear learning methods, such as Markov state models (MSMs), Hidden Markov Models and Koopman models, as well as kernel and deep learning approaches such as VAMPnets and deep MSMs. The library is largely compatible with scikit-learn, having a range of Estimator classes for these different models, but in contrast to scikit-learn also provides deep Model classes, e.g. in the case of an MSM, which provide a multitude of analysis methods to compute interesting thermodynamic, kinetic and dynamical quantities, such as free energies, relaxation times and transition paths. The library is designed for ease of use but also easily maintainable and extensible code. In this paper we introduce the main features and structure of the deeptime software.
Machine Learning Implicit Solvation for Molecular Dynamics
Jun 14, 2021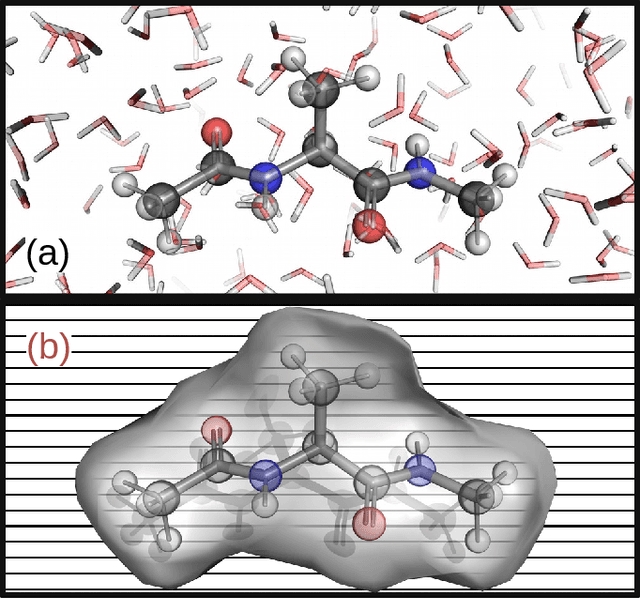
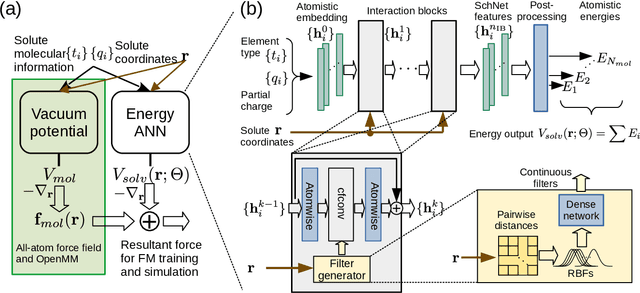
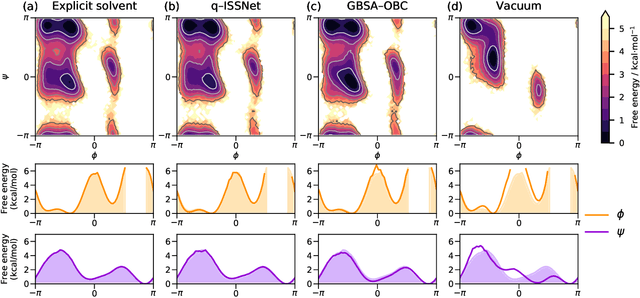
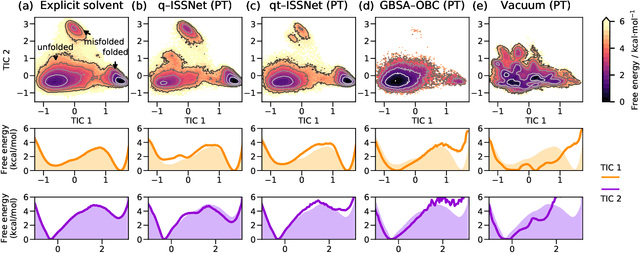
Accurate modeling of the solvent environment for biological molecules is crucial for computational biology and drug design. A popular approach to achieve long simulation time scales for large system sizes is to incorporate the effect of the solvent in a mean-field fashion with implicit solvent models. However, a challenge with existing implicit solvent models is that they often lack accuracy or certain physical properties compared to explicit solvent models, as the many-body effects of the neglected solvent molecules is difficult to model as a mean field. Here, we leverage machine learning (ML) and multi-scale coarse graining (CG) in order to learn implicit solvent models that can approximate the energetic and thermodynamic properties of a given explicit solvent model with arbitrary accuracy, given enough training data. Following the previous ML--CG models CGnet and CGSchnet, we introduce ISSNet, a graph neural network, to model the implicit solvent potential of mean force. ISSNet can learn from explicit solvent simulation data and be readily applied to MD simulations. We compare the solute conformational distributions under different solvation treatments for two peptide systems. The results indicate that ISSNet models can outperform widely-used generalized Born and surface area models in reproducing the thermodynamics of small protein systems with respect to explicit solvent. The success of this novel method demonstrates the potential benefit of applying machine learning methods in accurate modeling of solvent effects for in silico research and biomedical applications.
Coarse Graining Molecular Dynamics with Graph Neural Networks
Aug 21, 2020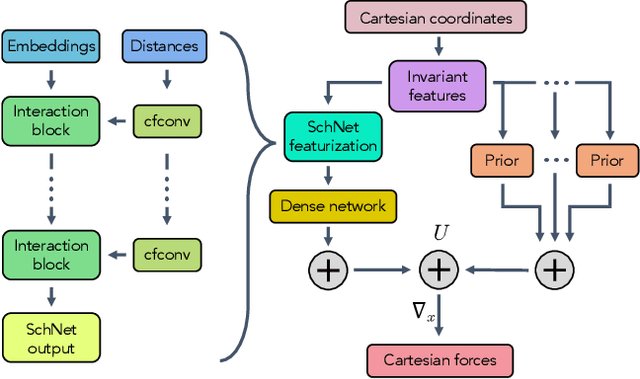
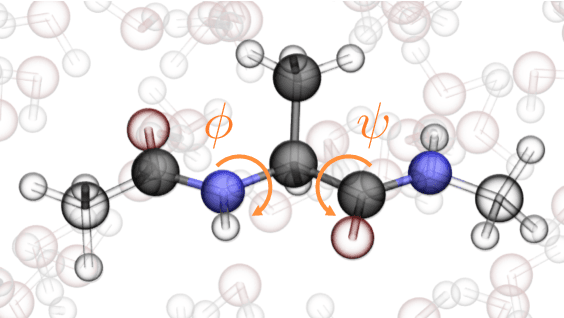
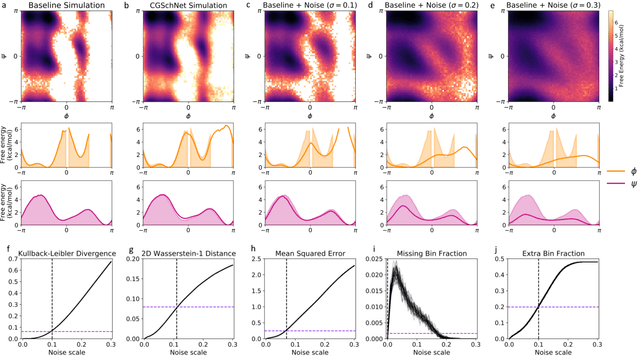
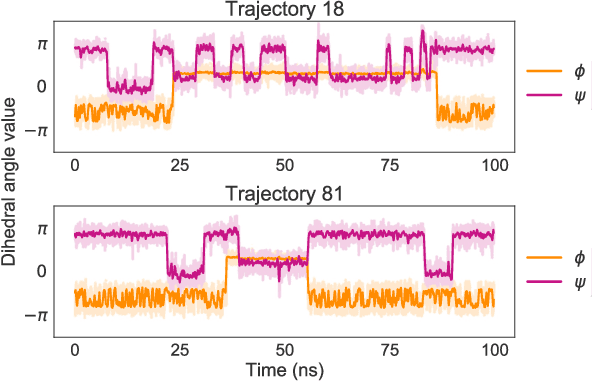
Coarse graining enables the investigation of molecular dynamics for larger systems and at longer timescales than is possible at atomic resolution. However, a coarse graining model must be formulated such that the conclusions we draw from it are consistent with the conclusions we would draw from a model at a finer level of detail. It has been proven that a force matching scheme defines a thermodynamically consistent coarse-grained model for an atomistic system in the variational limit. Wang et al. [ACS Cent. Sci. 5, 755 (2019)] demonstrated that the existence of such a variational limit enables the use of a supervised machine learning framework to generate a coarse-grained force field, which can then be used for simulation in the coarse-grained space. Their framework, however, requires the manual input of molecular features upon which to machine learn the force field. In the present contribution, we build upon the advance of Wang et al.and introduce a hybrid architecture for the machine learning of coarse-grained force fields that learns their own features via a subnetwork that leverages continuous filter convolutions on a graph neural network architecture. We demonstrate that this framework succeeds at reproducing the thermodynamics for small biomolecular systems. Since the learned molecular representations are inherently transferable, the architecture presented here sets the stage for the development of machine-learned, coarse-grained force fields that are transferable across molecular systems.
Kernel canonical correlation analysis approximates operators for the detection of coherent structures in dynamical data
Apr 16, 2019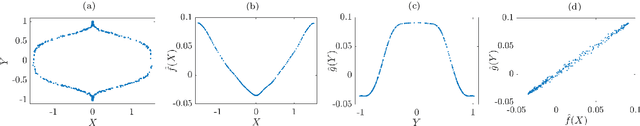
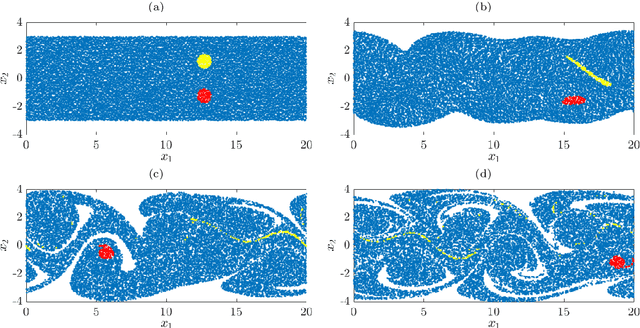
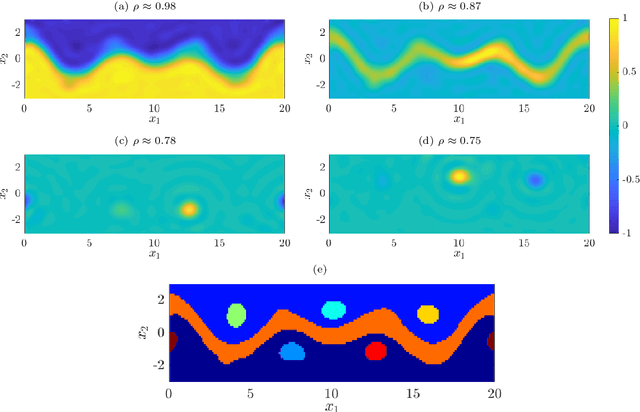
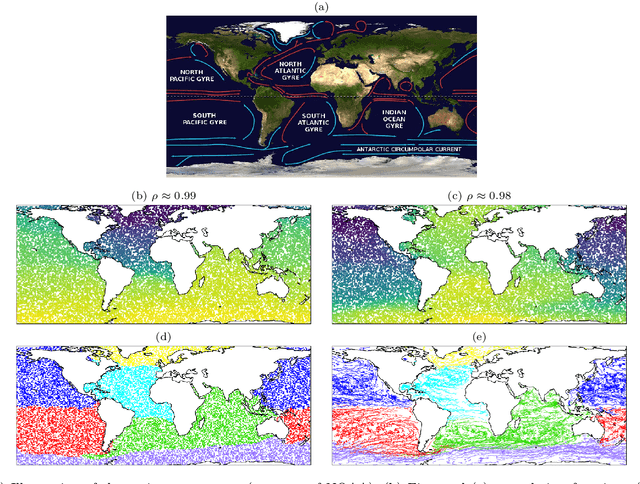
We illustrate relationships between classical kernel-based dimensionality reduction techniques and eigendecompositions of empirical estimates of reproducing kernel Hilbert space (RKHS) operators associated with dynamical systems. In particular, we show that kernel canonical correlation analysis (CCA) can be interpreted in terms of kernel transfer operators and that coherent sets of particle trajectories can be computed by applying kernel CCA to Lagrangian data. We demonstrate the efficiency of this approach with several examples, namely the well-known Bickley jet, ocean drifter data, and a molecular dynamics problem with a time-dependent potential. Furthermore, we propose a straightforward generalization of dynamic mode decomposition (DMD) called coherent mode decomposition (CMD).
Variational Selection of Features for Molecular Kinetics
Nov 28, 2018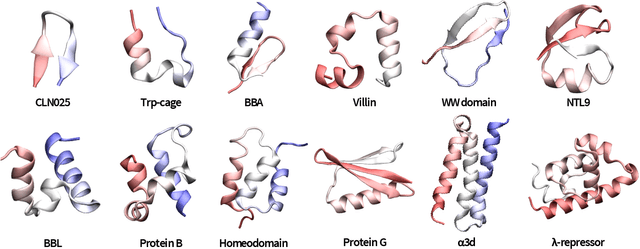
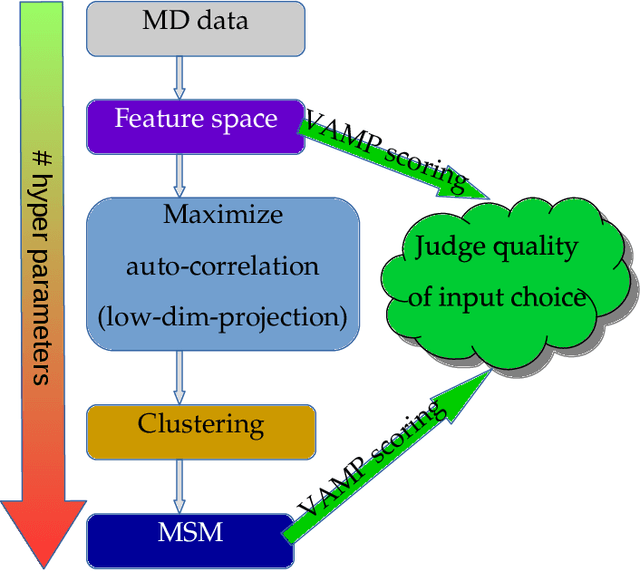
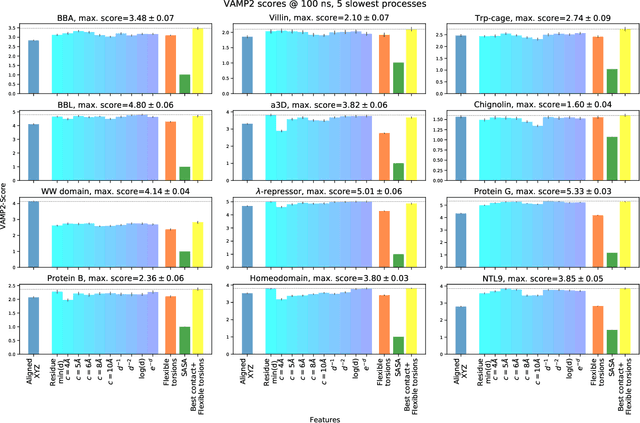
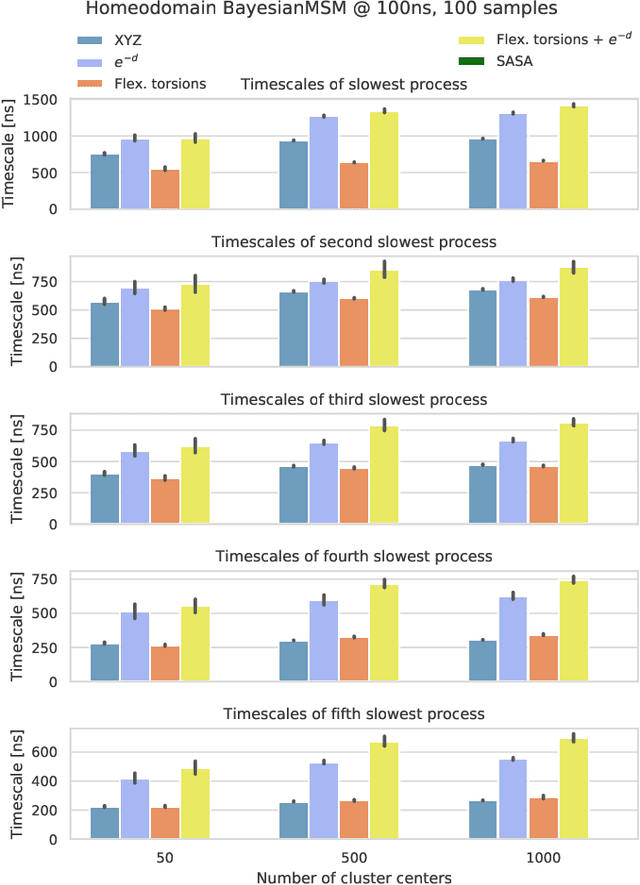
The modeling of atomistic biomolecular simulations using kinetic models such as Markov state models (MSMs) has had many notable algorithmic advances in recent years. The variational principle has opened the door for a nearly fully automated toolkit for selecting models that predict the long-time kinetics from molecular dynamics simulations. However, one yet-unoptimized step of the pipeline involves choosing the features, or collective variables, from which the model should be constructed. In order to build intuitive models, these collective variables are often sought to be interpretable and familiar features, such as torsional angles or contact distances in a protein structure. However, previous approaches for evaluating the chosen features rely on constructing a full MSM, which in turn requires additional hyperparameters to be chosen, and hence leads to a computationally expensive framework. Here, we present a method to optimize the feature choice directly, without requiring the construction of the final kinetic model. We demonstrate our rigorous preprocessing algorithm on a canonical set of twelve fast-folding protein simulations, and show that our procedure leads to more efficient model selection.
PotentialNet for Molecular Property Prediction
Oct 22, 2018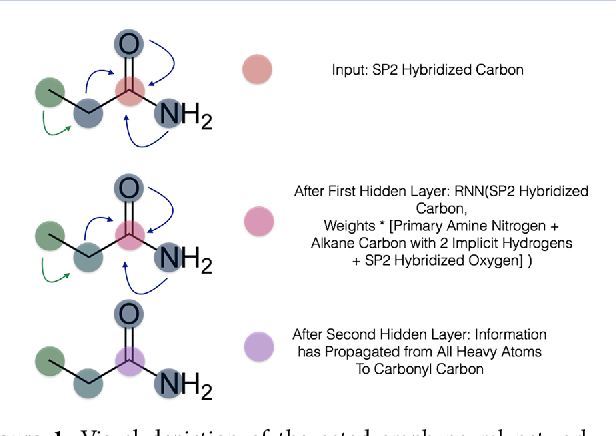
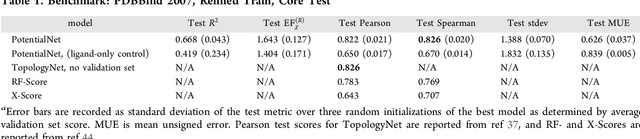
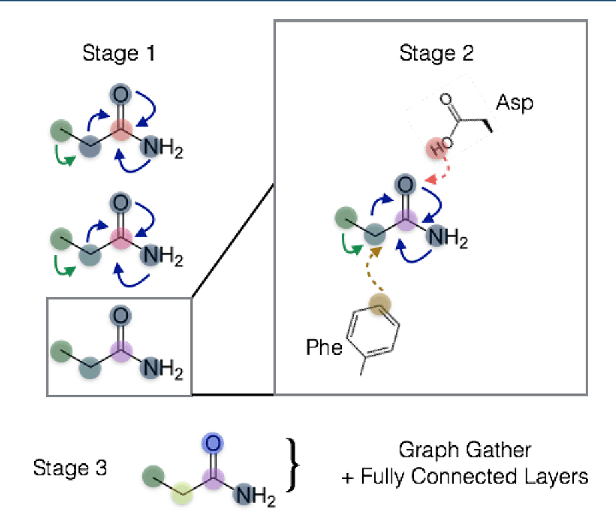
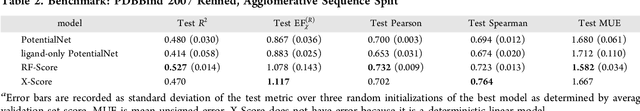
The arc of drug discovery entails a multiparameter optimization problem spanning vast length scales. They key parameters range from solubility (angstroms) to protein-ligand binding (nanometers) to in vivo toxicity (meters). Through feature learning---instead of feature engineering---deep neural networks promise to outperform both traditional physics-based and knowledge-based machine learning models for predicting molecular properties pertinent to drug discovery. To this end, we present the PotentialNet family of graph convolutions. These models are specifically designed for and achieve state-of-the-art performance for protein-ligand binding affinity. We further validate these deep neural networks by setting new standards of performance in several ligand-based tasks. In parallel, we introduce a new metric, the Regression Enrichment Factor $EF_\chi^{(R)}$, to measure the early enrichment of computational models for chemical data. Finally, we introduce a cross-validation strategy based on structural homology clustering that can more accurately measure model generalizability, which crucially distinguishes the aims of machine learning for drug discovery from standard machine learning tasks.
Simultaneous Coherent Structure Coloring facilitates interpretable clustering of scientific data by amplifying dissimilarity
Sep 03, 2018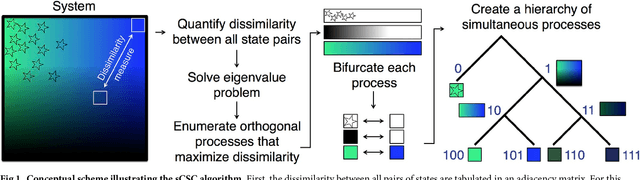
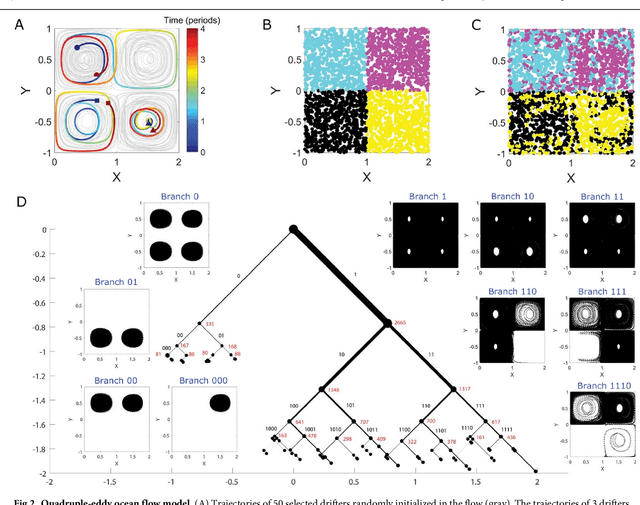
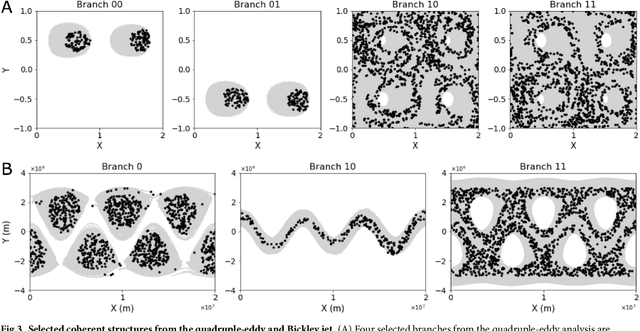
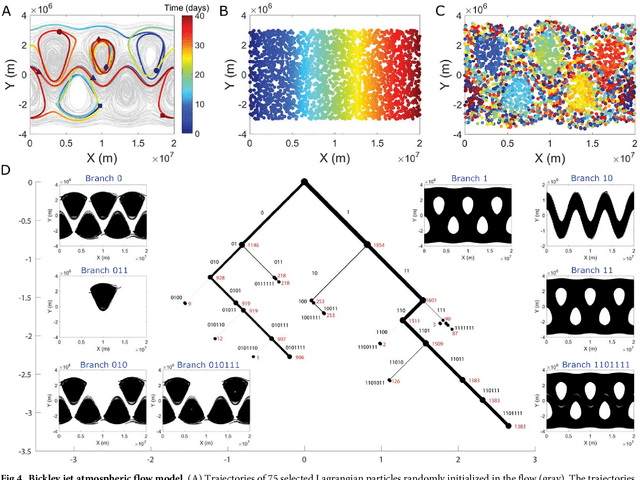
The clustering of data into physically meaningful subsets often requires assumptions regarding the number, size, or shape of the subgroups. Here, we present a new method, simultaneous Coherent Structure Coloring (sCSC), which accomplishes the task of unsupervised clustering without a priori guidance regarding the underlying structure of the data. sCSC performs a sequence of binary splittings on the dataset such that the most dissimilar data points are required to be in separate clusters. To achieve this, we obtain a set of orthogonal coordinates along which dissimilarity in the dataset is maximized from a generalized eigenvalue problem based on the pairwise dissimilarity between the data points to be clustered. This sequence of bifurcations produces a binary tree representation of the system, from which the number of clusters in the data and their interrelationships naturally emerge. To illustrate the effectiveness of the method in the absence of a priori assumptions we apply it to two exemplary problems in fluid dynamics. Then, we illustrate its capacity for interpretability using a high-dimensional protein folding simulation dataset. While we restrict our examples to dynamical physical systems in this work, we anticipate straightforward translation to other fields where existing analysis tools require ad hoc assumptions on the data structure, lack the interpretability of the present method, or in which the underlying processes are less accessible, such as genomics and neuroscience.
Unsupervised learning of dynamical and molecular similarity using variance minimization
Dec 20, 2017

In this report, we present an unsupervised machine learning method for determining groups of molecular systems according to similarity in their dynamics or structures using Ward's minimum variance objective function. We first apply the minimum variance clustering to a set of simulated tripeptides using the information theoretic Jensen-Shannon divergence between Markovian transition matrices in order to gain insight into how point mutations affect protein dynamics. Then, we extend the method to partition two chemoinformatic datasets according to structural similarity to motivate a train/validation/test split for supervised learning that avoids overfitting.