 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSemantically Conditioned Diffusion Models for Cerebral DSA Synthesis
Feb 12, 2026Digital subtraction angiography (DSA) plays a central role in the diagnosis and treatment of cerebrovascular disease, yet its invasive nature and high acquisition cost severely limit large-scale data collection and public data sharing. Therefore, we developed a semantically conditioned latent diffusion model (LDM) that synthesizes arterial-phase cerebral DSA frames under explicit control of anatomical circulation (anterior vs.\ posterior) and canonical C-arm positions. We curated a large single-centre DSA dataset of 99,349 frames and trained a conditional LDM using text embeddings that encoded anatomy and acquisition geometry. To assess clinical realism, four medical experts, including two neuroradiologists, one neurosurgeon, and one internal medicine expert, systematically rated 400 synthetic DSA images using a 5-grade Likert scale for evaluating proximal large, medium, and small peripheral vessels. The generated images achieved image-wise overall Likert scores ranging from 3.1 to 3.3, with high inter-rater reliability (ICC(2,k) = 0.80--0.87). Distributional similarity to real DSA frames was supported by a low median Fréchet inception distance (FID) of 15.27. Our results indicate that semantically controlled LDMs can produce realistic synthetic DSAs suitable for downstream algorithm development, research, and training.
AI Agents in Drug Discovery
Oct 31, 2025Artificial intelligence (AI) agents are emerging as transformative tools in drug discovery, with the ability to autonomously reason, act, and learn through complicated research workflows. Building on large language models (LLMs) coupled with perception, computation, action, and memory tools, these agentic AI systems could integrate diverse biomedical data, execute tasks, carry out experiments via robotic platforms, and iteratively refine hypotheses in closed loops. We provide a conceptual and technical overview of agentic AI architectures, ranging from ReAct and Reflection to Supervisor and Swarm systems, and illustrate their applications across key stages of drug discovery, including literature synthesis, toxicity prediction, automated protocol generation, small-molecule synthesis, drug repurposing, and end-to-end decision-making. To our knowledge, this represents the first comprehensive work to present real-world implementations and quantifiable impacts of agentic AI systems deployed in operational drug discovery settings. Early implementations demonstrate substantial gains in speed, reproducibility, and scalability, compressing workflows that once took months into hours while maintaining scientific traceability. We discuss the current challenges related to data heterogeneity, system reliability, privacy, and benchmarking, and outline future directions towards technology in support of science and translation.
Enhancing Traffic Accident Classifications: Application of NLP Methods for City Safety
Jun 11, 2025A comprehensive understanding of traffic accidents is essential for improving city safety and informing policy decisions. In this study, we analyze traffic incidents in Munich to identify patterns and characteristics that distinguish different types of accidents. The dataset consists of both structured tabular features, such as location, time, and weather conditions, as well as unstructured free-text descriptions detailing the circumstances of each accident. Each incident is categorized into one of seven predefined classes. To assess the reliability of these labels, we apply NLP methods, including topic modeling and few-shot learning, which reveal inconsistencies in the labeling process. These findings highlight potential ambiguities in accident classification and motivate a refined predictive approach. Building on these insights, we develop a classification model that achieves high accuracy in assigning accidents to their respective categories. Our results demonstrate that textual descriptions contain the most informative features for classification, while the inclusion of tabular data provides only marginal improvements. These findings emphasize the critical role of free-text data in accident analysis and highlight the potential of transformer-based models in improving classification reliability.
A Large-Scale Neutral Comparison Study of Survival Models on Low-Dimensional Data
Jun 06, 2024



This work presents the first large-scale neutral benchmark experiment focused on single-event, right-censored, low-dimensional survival data. Benchmark experiments are essential in methodological research to scientifically compare new and existing model classes through proper empirical evaluation. Existing benchmarks in the survival literature are often narrow in scope, focusing, for example, on high-dimensional data. Additionally, they may lack appropriate tuning or evaluation procedures, or are qualitative reviews, rather than quantitative comparisons. This comprehensive study aims to fill the gap by neutrally evaluating a broad range of methods and providing generalizable conclusions. We benchmark 18 models, ranging from classical statistical approaches to many common machine learning methods, on 32 publicly available datasets. The benchmark tunes for both a discrimination measure and a proper scoring rule to assess performance in different settings. Evaluating on 8 survival metrics, we assess discrimination, calibration, and overall predictive performance of the tested models. Using discrimination measures, we find that no method significantly outperforms the Cox model. However, (tuned) Accelerated Failure Time models were able to achieve significantly better results with respect to overall predictive performance as measured by the right-censored log-likelihood. Machine learning methods that performed comparably well include Oblique Random Survival Forests under discrimination, and Cox-based likelihood-boosting under overall predictive performance. We conclude that for predictive purposes in the standard survival analysis setting of low-dimensional, right-censored data, the Cox Proportional Hazards model remains a simple and robust method, sufficient for practitioners.
Training Survival Models using Scoring Rules
Mar 19, 2024



Survival Analysis provides critical insights for partially incomplete time-to-event data in various domains. It is also an important example of probabilistic machine learning. The probabilistic nature of the predictions can be exploited by using (proper) scoring rules in the model fitting process instead of likelihood-based optimization. Our proposal does so in a generic manner and can be used for a variety of model classes. We establish different parametric and non-parametric sub-frameworks that allow different degrees of flexibility. Incorporated into neural networks, it leads to a computationally efficient and scalable optimization routine, yielding state-of-the-art predictive performance. Finally, we show that using our framework, we can recover various parametric models and demonstrate that optimization works equally well when compared to likelihood-based methods.
Understanding Biology in the Age of Artificial Intelligence
Mar 06, 2024



Modern life sciences research is increasingly relying on artificial intelligence approaches to model biological systems, primarily centered around the use of machine learning (ML) models. Although ML is undeniably useful for identifying patterns in large, complex data sets, its widespread application in biological sciences represents a significant deviation from traditional methods of scientific inquiry. As such, the interplay between these models and scientific understanding in biology is a topic with important implications for the future of scientific research, yet it is a subject that has received little attention. Here, we draw from an epistemological toolkit to contextualize recent applications of ML in biological sciences under modern philosophical theories of understanding, identifying general principles that can guide the design and application of ML systems to model biological phenomena and advance scientific knowledge. We propose that conceptions of scientific understanding as information compression, qualitative intelligibility, and dependency relation modelling provide a useful framework for interpreting ML-mediated understanding of biological systems. Through a detailed analysis of two key application areas of ML in modern biological research - protein structure prediction and single cell RNA-sequencing - we explore how these features have thus far enabled ML systems to advance scientific understanding of their target phenomena, how they may guide the development of future ML models, and the key obstacles that remain in preventing ML from achieving its potential as a tool for biological discovery. Consideration of the epistemological features of ML applications in biology will improve the prospects of these methods to solve important problems and advance scientific understanding of living systems.
Evaluating machine learning models in non-standard settings: An overview and new findings
Oct 23, 2023



Estimating the generalization error (GE) of machine learning models is fundamental, with resampling methods being the most common approach. However, in non-standard settings, particularly those where observations are not independently and identically distributed, resampling using simple random data divisions may lead to biased GE estimates. This paper strives to present well-grounded guidelines for GE estimation in various such non-standard settings: clustered data, spatial data, unequal sampling probabilities, concept drift, and hierarchically structured outcomes. Our overview combines well-established methodologies with other existing methods that, to our knowledge, have not been frequently considered in these particular settings. A unifying principle among these techniques is that the test data used in each iteration of the resampling procedure should reflect the new observations to which the model will be applied, while the training data should be representative of the entire data set used to obtain the final model. Beyond providing an overview, we address literature gaps by conducting simulation studies. These studies assess the necessity of using GE-estimation methods tailored to the respective setting. Our findings corroborate the concern that standard resampling methods often yield biased GE estimates in non-standard settings, underscoring the importance of tailored GE estimation.
Deep Learning for Survival Analysis: A Review
May 24, 2023The influx of deep learning (DL) techniques into the field of survival analysis in recent years, coupled with the increasing availability of high-dimensional omics data and unstructured data like images or text, has led to substantial methodological progress; for instance, learning from such high-dimensional or unstructured data. Numerous modern DL-based survival methods have been developed since the mid-2010s; however, they often address only a small subset of scenarios in the time-to-event data setting - e.g., single-risk right-censored survival tasks - and neglect to incorporate more complex (and common) settings. Partially, this is due to a lack of exchange between experts in the respective fields. In this work, we provide a comprehensive systematic review of DL-based methods for time-to-event analysis, characterizing them according to both survival- and DL-related attributes. In doing so, we hope to provide a helpful overview to practitioners who are interested in DL techniques applicable to their specific use case as well as to enable researchers from both fields to identify directions for future investigation. We provide a detailed characterization of the methods included in this review as an open-source, interactive table: https://survival-org.github.io/DL4Survival. As this research area is advancing rapidly, we encourage the research community to contribute to keeping the information up to date.
Conditional Neural Processes for Molecules
Oct 17, 2022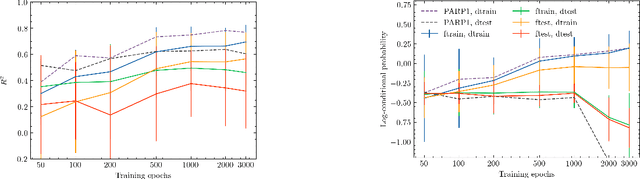
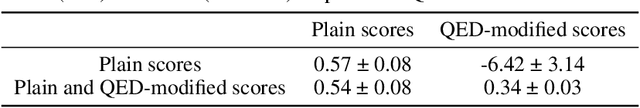

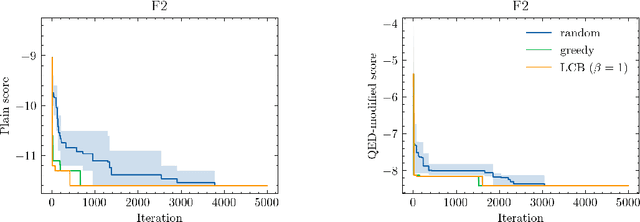
Neural processes (NPs) are models for transfer learning with properties reminiscent of Gaussian Processes (GPs). They are adept at modelling data consisting of few observations of many related functions on the same input space and are trained by minimizing a variational objective, which is computationally much less expensive than the Bayesian updating required by GPs. So far, most studies of NPs have focused on low-dimensional datasets which are not representative of realistic transfer learning tasks. Drug discovery is one application area that is characterized by datasets consisting of many chemical properties or functions which are sparsely observed, yet depend on shared features or representations of the molecular inputs. This paper applies the conditional neural process (CNP) to DOCKSTRING, a dataset of docking scores for benchmarking ML models. CNPs show competitive performance in few-shot learning tasks relative to supervised learning baselines common in QSAR modelling, as well as an alternative model for transfer learning based on pre-training and refining neural network regressors. We present a Bayesian optimization experiment which showcases the probabilistic nature of CNPs and discuss shortcomings of the model in uncertainty quantification.
Heterogeneous Treatment Effect Estimation for Observational Data using Model-based Forests
Oct 06, 2022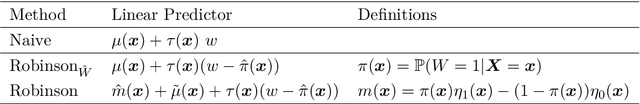
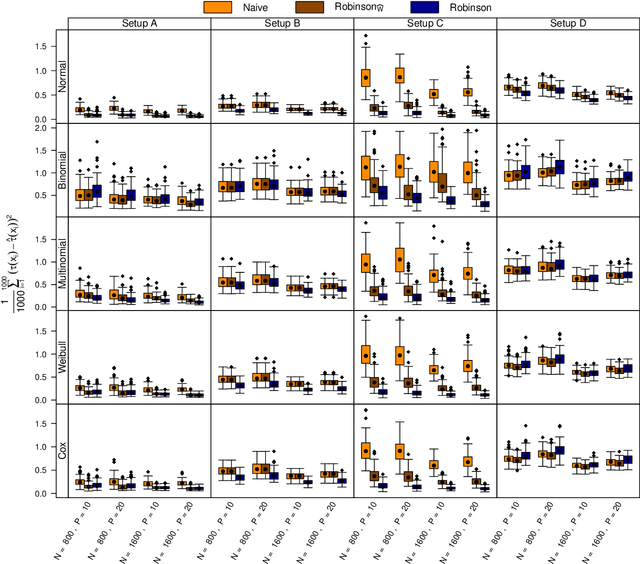
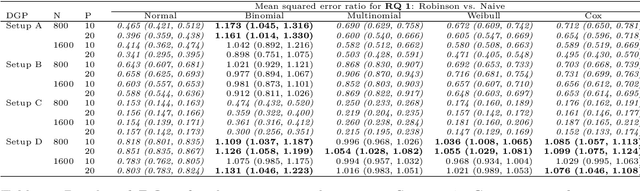
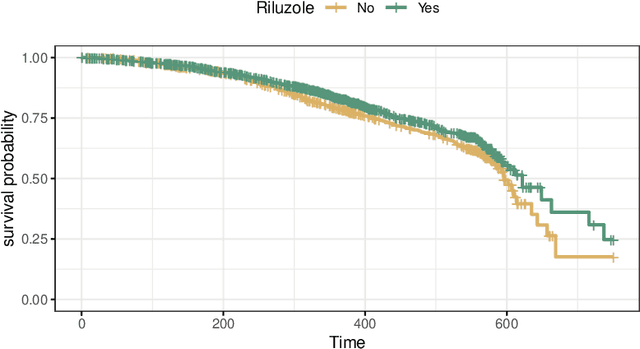
The estimation of heterogeneous treatment effects (HTEs) has attracted considerable interest in many disciplines, most prominently in medicine and economics. Contemporary research has so far primarily focused on continuous and binary responses where HTEs are traditionally estimated by a linear model, which allows the estimation of constant or heterogeneous effects even under certain model misspecifications. More complex models for survival, count, or ordinal outcomes require stricter assumptions to reliably estimate the treatment effect. Most importantly, the noncollapsibility issue necessitates the joint estimation of treatment and prognostic effects. Model-based forests allow simultaneous estimation of covariate-dependent treatment and prognostic effects, but only for randomized trials. In this paper, we propose modifications to model-based forests to address the confounding issue in observational data. In particular, we evaluate an orthogonalization strategy originally proposed by Robinson (1988, Econometrica) in the context of model-based forests targeting HTE estimation in generalized linear models and transformation models. We found that this strategy reduces confounding effects in a simulated study with various outcome distributions. We demonstrate the practical aspects of HTE estimation for survival and ordinal outcomes by an assessment of the potentially heterogeneous effect of Riluzole on the progress of Amyotrophic Lateral Sclerosis.