 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeHelixFold-Single: MSA-free Protein Structure Prediction by Using Protein Language Model as an Alternative
Paper and Code
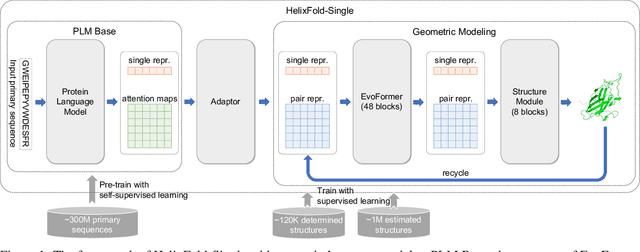

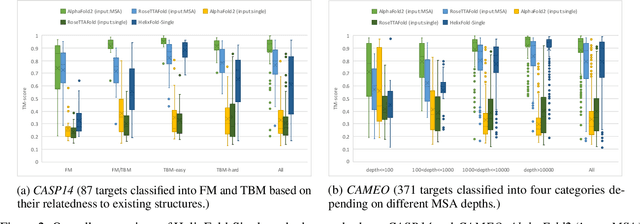
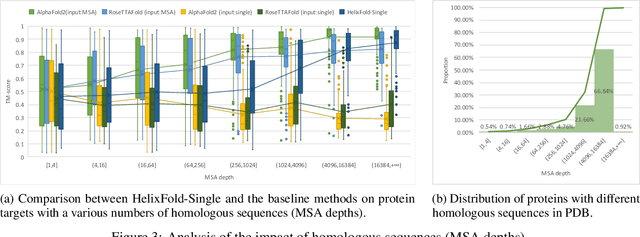
AI-based protein structure prediction pipelines, such as AlphaFold2, have achieved near-experimental accuracy. These advanced pipelines mainly rely on Multiple Sequence Alignments (MSAs) as inputs to learn the co-evolution information from the homologous sequences. Nonetheless, searching MSAs from protein databases is time-consuming, usually taking dozens of minutes. Consequently, we attempt to explore the limits of fast protein structure prediction by using only primary sequences of proteins. HelixFold-Single is proposed to combine a large-scale protein language model with the superior geometric learning capability of AlphaFold2. Our proposed method, HelixFold-Single, first pre-trains a large-scale protein language model (PLM) with thousands of millions of primary sequences utilizing the self-supervised learning paradigm, which will be used as an alternative to MSAs for learning the co-evolution information. Then, by combining the pre-trained PLM and the essential components of AlphaFold2, we obtain an end-to-end differentiable model to predict the 3D coordinates of atoms from only the primary sequence. HelixFold-Single is validated in datasets CASP14 and CAMEO, achieving competitive accuracy with the MSA-based methods on the targets with large homologous families. Furthermore, HelixFold-Single consumes much less time than the mainstream pipelines for protein structure prediction, demonstrating its potential in tasks requiring many predictions. The code of HelixFold-Single is available at https://github.com/PaddlePaddle/PaddleHelix/tree/dev/apps/protein_folding/helixfold-single, and we also provide stable web services on https://paddlehelix.baidu.com/app/drug/protein-single/forecast.