 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDual-view Molecule Pre-training
Paper and Code
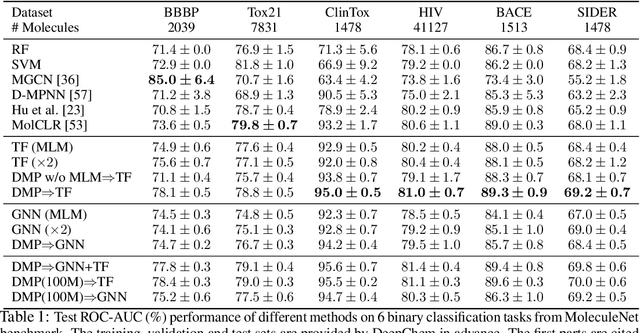
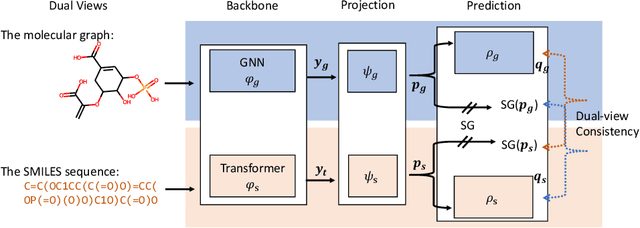
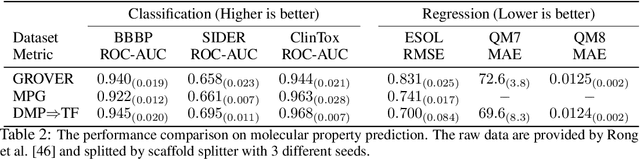
Inspired by its success in natural language processing and computer vision, pre-training has attracted substantial attention in cheminformatics and bioinformatics, especially for molecule based tasks. A molecule can be represented by either a graph (where atoms are connected by bonds) or a SMILES sequence (where depth-first-search is applied to the molecular graph with specific rules). Existing works on molecule pre-training use either graph representations only or SMILES representations only. In this work, we propose to leverage both the representations and design a new pre-training algorithm, dual-view molecule pre-training (briefly, DMP), that can effectively combine the strengths of both types of molecule representations. The model of DMP consists of two branches: a Transformer branch that takes the SMILES sequence of a molecule as input, and a GNN branch that takes a molecular graph as input. The training of DMP contains three tasks: (1) predicting masked tokens in a SMILES sequence by the Transformer branch, (2) predicting masked atoms in a molecular graph by the GNN branch, and (3) maximizing the consistency between the two high-level representations output by the Transformer and GNN branches separately. After pre-training, we can use either the Transformer branch (this one is recommended according to empirical results), the GNN branch, or both for downstream tasks. DMP is tested on nine molecular property prediction tasks and achieves state-of-the-art performances on seven of them. Furthermore, we test DMP on three retrosynthesis tasks and achieve state-of-the-result on the USPTO-full dataset. Our code will be released soon.